ChemAxon Extended SMILES and SMARTS (CXSMILES and CXSMARTS)
ChemAxon Extended SMILES/SMARTS is used for storing special features of the molecules after the SMILES string. Any information can be stored after the SMILES string if it is separated by space or tab characters as the SMILES parsers ignore them or use them as comment. The extended features are stored in the following format:
SMILES_String |<feature1>,<feature2>,...|ChemAxon's extended SMILES/SMARTS does not contain non-ASCII characters, they are escaped in the usual form, "&#n;", with their character code, n. The ASCII characters ',', ';', '|', '{', '}' and ':' in Data S-group information are also escaped in this way. Moreover, the symbols '$', ';', '|', '{', '}' between dollar signs (see Atom labels / aliases / values) are coded in the above mentioned way as well.
The extended feature description is economic. If some feature is missing in the molecule, then the corresponding special characters are not written (e.g.: If the atoms of the molecule has no alias strings at all, no "$" and ";" characters are written.). Moreover, if no feature of the molecule to be written, the extended feature field is omitted.
Please note that the SMILES string part generated in CXSMILES format is not always the same as the one generated by smiles output. For example, in case of Ferrocene the coordinate bonds are not exported to plain SMILES ([Fe].c1cccc1.c1cccc1), but they appear in the CXSMILES (c12c3c4c5c1[Fe]23451234c5c1c2c3c45 |C:4.5,0.6,1.7,2.8,3.9,7.12,6.10,9.16,10.18,8.14|).
In extended smiles export the following additional features are exported:
Aromatic atoms
All aromatic atom are exported with lowercase letter in the SMILES string part.
E.g. aromatic Boron is written with lowercase letter: b1ccccc1.
Atom labels / aliases / values
Atom labels / aliases are written between "$" characters each label is separated by ";" characters.
Atom values are written after "$_AV:" separated by semicolon characters and closed with "$" tag. Special characters are escaped.
Link nodes
The link node atom indexes are written after "LN:" followed by a colon character, the minimum repetitions, maximum repetitions, the node first and second outer atom indexes separated by ".". If the link node has only two connections, then the first and second outer atom indexes are obvious, so they are omitted. The link nodes separated by commas.
Example:
LN:1:1.5.3.0,6:1.2.7.5,9:1.10.10.8
Codename: cxsmiles, cxsmarts
Following features included
See also
Atomic coordinates
The atomic coordinates are written between parentheses. Each atomic coordinate triplet (x, y, z) is separated by semicolon, and the x y z coordinates are separated by commas. Zero coordinates are omitted.
Note: The CIS/TRANS information is redundant in this case. It is specified in the SMILES string and also in the atomic coordinates. The atomic coordinates has priority over the SMILES string.
Atom properties
Atom properties are exported to CXSMILES and CXSMARTS after the keyword 'atomProp' at the extended part. Every property is exported separately with the following rule:
-
first comes the atom index
-
second is the property key (after a dot)
-
last is the property value (after a dot)
The properties are separated with colons. The end of the atom property block is marked with a comma. If the atom has a non-string property, an exception is thrown.
Example:
-
CNC |atomprop:0.key1.value1:0.key2.value2:1.key3.value3| // The 0th atom has two properties and the 1 indexed atom has one.
Pseudo Atoms
Pseudo atoms can be exported to extended CXSMILES/CXSMARTS. They are described in the alias part as "pseudo_p", where pseudo is the value of the pseudo atom.
Example:
|
CCCC* |$;;;;pseudo_p$| |
|
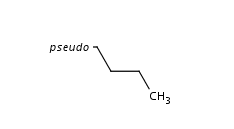
Special Atoms
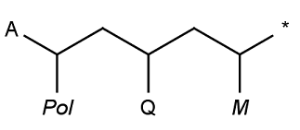
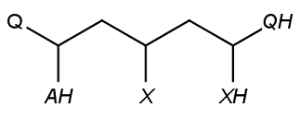
Special atoms AH, QH, M, MH, X, XH and Pol, are exported to cxsmiles/cxsmarts as pseudo atoms, i.e. AH_p, QH_p, M_p, MH_p, X_p, XH_p, and Pol_p, respectively. Special atoms Q and star are exported as Q_e and star_e, respectively. Special atom A can be handled by SMILES export, therefore it is not written to the alias part of the cxsmiles/cxsmarts.
Examples:
|
*C(*)CC(*)CC(*)* |$;;Pol_p;;;Q_e;;;star_e;M_p$| |
|
|
*C(*)CC(*)CC(*)* |$Q_e;;AH_p;;;X_p;;;QH_p;XH_p$| |
|
Radical numbers
Atom indexes with
-
monovalent radical center are written after "^1:",
-
divalent radical center are written after "^2:",
-
divalent singlet radical center are written after "^3:",
-
divalent triplet radical center are written after "^4:",
-
trivalent radical center are written after "^5:",
-
trivalent doublet radical center are written after "^6:",
-
trivalent quartet radical center are written after "^7:",
characters separated by commas.
Lone electron pairs
The indexes of the atoms having bond connected lone electron pairs are written after "LP:".
The indexes of the atoms followed by a colon character and the number of explicit lone electron pairs are written after "lp:".
(See live example.)
Example: "LP:1,lp:0:1,2:2"
Coordinate and Hydrogen bonds
The atom index of the first atom in the coordinate bond is written after "C:" followed by a dot character and the coordinate bond index. The coordinate bonds are separated by commas.
In the smiles part of CXSMILES the atom-to-atom coordinate bonds are represented by single bonds, which are corrected according to the C information at the extended part. Hydrogen bonds exported in the same format after "H:".
Example:
-
Coordinate bond: "C:0.0,2.1"
-
Hydrogen bond: "CO(C)[H]N1C=CC=C1 |c:5,7,H:3.2| "
Molecule absolute stereoconfiguration
(For detailed description see the Stereochemistry section of the Query guide in JChem Base.)
The relative stereoconfiguration is stored as "r". If a reaction contains components with absolute and relative stereo the indexes of the fragments with relative configuration is written. The absolute stereoconfiguration is the default, which is not marked. (Absolute stereoconfiguration known also as "Chiral flag" in MDL molfiles. )
Example: "r:2,4,5"
Enhanced stereochemical representation
(For detailed description see the Stereochemistry section of the Query guide in JChem Base.)
The following stereochemical group types are stored:
-
Absolute stereo group type.
a:<atomindex>,<atomindex>... -
OR stereo group type.
o<group>:<atomindex>,< atomindex>... -
AND stereo group type.
&<group>:<atomindex>,< atomindex>...
Single "Up or Down" (Wiggly), UP and DOWN bonds
Atom indexes relating to wiggly bonds are written after "w:" followed by a dot character and the wiggly bond index. The wiggly bonds are separated by commas.
If atomic coordinates are also exported, then UP bonds are written after "wU:" DOWN bonds are written after "wD:" in a similar way to wiggly bond export.
CIS, TRANS, UNSPEC bond info for double bonds in rings
Bond indexes of the double bonds in SSSR are written.
The bond stereo information is generated as the following: the double bond has the representation a1-a2=a3-a4, where
-
a1 is the smallest atom index of the generated smiles connected to a2
-
a2 is the double bond smaller atom index in the generated smiles
-
a3 is the double bond larger atom index in the generated smiles
-
a4 is the smallest atom index of the generated smiles connected to a3
The CIS double bond indexes are written after "c:", the TRANS double bond indexes are written after "t:", the double bond indexes with UNSPEC flag are written after "ctu:".
Local parity information
Atom indexes with local ODD parity are written after "@:", while atom indexes with local EVEN parity are written after "@@:" characters separated by commas.
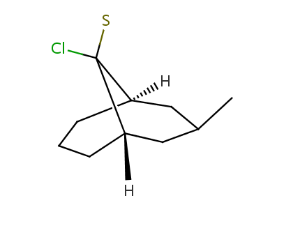
Local bicyclo-alkane stereo information (local syn/anti, endo/exo representation)
For each ligand connected to non-bridgehead atoms of bicyclo-alkanes, if they are in a syn/anti or endo/exo position (ligand is not in the plane of the bridge to which it is connected), their relative position in the ring system is stored by their position in relation to the bridges to which they are not connected. Bridges are identified by the indexes of the contained atoms: higher bridge is the one with the highest atom index, the other is the lower bridge. The ligand's position can be:
-
towards higher bridge (THB), if it lies towards the higher bridge regarding the plane of the connected bridge
-
towards lower bridge (TLB), if it lies towards the lower bridge regarding the plane of the connected bridge
-
towards either bridge (TEB), if its position is not determined, it can lie towards both of the bridges (it is connected by a wiggly bond)
The order of the stored information: at first each THB, than each TLB, in the end each TEB description is written in the following form:
-
"THB", followed by ":", than for each THB ligand as follows:
-
index of the ligand atom for which bicyclo stereo description is stored, followed by ":"
-
index of the ring atom in the bicyclo-alkane to which the ligand is connected (connection atom), followed by ":"
-
list of indexes in the lower bridge, separated by "." and followed by ":"
-
list of indexes in the higher bridge, separated by "."
-
stereo description of separate ligands are separated by ","
-
-
the same lists are present for THB and TEB atoms, separated by ",".
If a ligand has more than one stereo information (e.g. it is connected to more than one bicyclo-alkanes) then it appears twice in the list with the adequate stereo description.
Examples:
[H][C@]12CCC[C@]([H])(CC(C)C1)C2(S)Cl |r,TLB:13:11:2.4.3:7.10.8,THB:12:11:2.4.3:7.10.8,9:8:11:2.4.3| |
|
[H][C@]12COC[C@]([H])(C[C@@H](CCCC)C1)[C@@]2(Cl)C1CCCC1 |r,TLB:15:14:2.4.3:7.13.8,THB:16:14:2.4.3:7.13.8,9:8:14:2.4.3| |
|
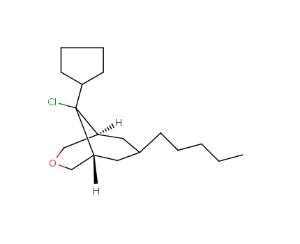
Fragment grouping
Fragment level grouping of reactant, agent and product fragments Grouped fragment indexes are written after "f:" in the following format:
-
Connected groups are separated by ",".
-
A connected group is a "." separated list of fragment indexes.
Example: "f:0.1,5.6"
Multicenter S-groups
The Multicenter atom indexes written after "m:" followed by a colon character and the indexes of the atoms which forms the given S-group separated by ".". The S-groups are separated by commas.
Example:
m:0:7.6.5.4.3,2:12.11.10.9.8,C:0.0,2.1
Data S-group information
Atomic indexes in the Data S-group are written after "SgD:" followed by field name, data value, query operator, unit, tag and coordinates in parenthesis if necessary, separated by colon characters. The field values with special characters are escaped. If atomic coordinates are exported (with option c ) (-1) is used in the coordinate field for Data S-group attached to the atoms.
Example:
SgD:3,2,1,0:name:data:like:unit:t:(-1)
Polymer S-groups
Each S-group exported after "Sg:" in fields separated by a colon. Fields are:
-
Sgroup type keyword. Valid keywords are:
Keyword
S-group Type
n
SRU
mon
monomer
mer
mer
co
copolymer
xl
crosslink
mod
modification
mix
mixture
f
formulation
any
anypolimer
gen
generic
c
component
grf
graft
alt
alternating copolymer
ran
random copolymer
blk
block copolymer
-
Atom indexes separated with commas.
-
Subscript of the S-group. If the subscript equals the keyword of the S-group this field can be empty. Escaped field.
-
Superscript of the S-group. In the superscript only connectivity and flip information is allowed. This field can be empty. Escaped field.
-
Head crossing bond indexes. The indexes of bonds that share a common bracket in case of ladder-type polymers. This field can be empty.
-
Tail crossing bond indexes. The indexes of bonds that share a common bracket in case of ladder-type polymers. This field can be empty.
-
If the export option c is present then bracket orientation, bracket type followed by the coordinates (4 pair, separated with commas). Bracket orientation can be s or d (single or double), bracket type can be b,c,r,s for braces, chevrons, round and square, respectively. The brackets are written between parentheses and separated with semicolons.
A colon is needed after the last non-empty field.
If one needs to retain not only the chemically relevant information, but the whole structure (as drawn), then the c export option should be used.
Examples:
CCCC |Sg:gen:0,1,2:|
CCCC |Sg:n:0,1,2:3-6:eu|
*CC(*)C(*)N* |$star_e;;;star_e;;star_e;;star_e$,Sg:n:6,1,2,4::hh,f:6,0,:4,2,|
C1=CC=CC=C1 |c:0,2,4,(-4.62,1.05,;-3.29,.28,;-3.29,-1.27,;-4.62,-2.04,;-5.95,-1.27,;-5.95,.28,),Sg:mon:0,5,4,3,2,1:::::(d,s,-7.03,2.12,-2.21,2.12,-2.21,-3.11,-7.03,-3.11,)|
S-group hierarchy
Parent-child relationship of the sgroups is described with the "SgH" tag.
The structure of the SgH tag is the following:
SgH:parentSgroupIndex1:childSgroupIndex1.childSgroupIndex2,parentSgroupIndex2:childSgroupIndex1
The indexes of the S-groups come from the order in they are written in the CXSMILES string, i.e. the first sgroup has the index 0, the second has 1, and so on. This includes datasgroups and polymer sgroups as well. Examples:
CC(N)C=O |Sg:gen:0::,Sg:mon:1,2,4,0,3::,SgH:1:0| // A monomer sgroup contains all 5 atoms, and it contains the generic sgroup with 1 atom.
C1CCCCC1 |SgD:0,1,2,3,4,5:f:34::::,Sg:mon:0,1,2,3,4,5::,SgH:1:0| // A monomer sgroup contains all the atoms, and it contains the datasgroup too.
C.C |SgD:1::::::,SgD:0,1::::::,SgD:0::::::,SgD:0::::::,Sg:gen:0::,Sg:gen:1::,Sg:gen:1::,SgH:5:6,6:0,2:4.3| // A more difficult example with multiple sgroup relations.
S-group attachment point information
S-group attachment point informations are not handled by CXSMILES or CXSMARTS.
R-groups
R-group information can be exported to extended CXSMILES/CXSMARTS. R-groups in the molecule is exported to ANY atom in the SMILES part, they are described in the alias part as "_Rn". Rgroup descriptions (molecules) are enumerated also in the extended part after "RG" followed by a colon character.
-
The R-group number is written after "_R" followed by "=" (e.g. _R1=)
-
The R-group description is written as CXSMILES/CXSMARTS in braces "{}"
-
Members of the same R-group are separated by commas
-
Different R-groups are separated by commas.
Examples
C1O[*]CO[*]1 |$;;_R2;;;_R1$,RG:_R1={C},{N},_R2={C},{N}|Cl[*](Br)I |$;_R1;;$,RG:_R1={*CCCC(C*)CC* |$_AP3;;;;;;_AP2;;;_AP1$|},{*CCCN(C*)CC* |$_AP3;;;;;;_AP2;;;_AP1$|},LO:1:0.2.3|
R-logic
R-logic is exported along with the R-group information. It is indicated by the LOG tag, which includes the list of R-logics for the R-groups. The list items are separated by dots. One item consits of the R-logic properties separated by semicolons: identifier of an other rgroup which is after the 'then' part of the R-logic (e.g. 'if R1>0 then R2'), the restH property ('H' if set, empty if not) and the R-logic range. If there is no R-logic specified for an R-group, then it is not included in the list.
Example:
[*]C1CCCCC1[*] |$_R1;;;;;;;_R2$,RG:_R1={CCC},_R2={N},LOG={_R1:;;>0._R2:_R1;H;0,1}| R-group attachment point information
The R-group attachment point is written explicitly as ANY atom into the SMILES string. The order of attachment point is written as alias string (see above) after "_AP" separated by semicolon characters. Before version 5.4 only two attachment point type was supported, the attachment point was not exported to the SMILES string explicitly. In the extended part the atomic indexes of the attachment points written after "AP_x:" format was used, where x denoted attachment type 1, 2 or 3 for attachment points 1, 2 or both.
Example:
-
C[C@H](N*)C(*)=O |$;;;_AP1;;_AP2;$|
-
before version 5.4: C[C@H]([NH])[C]=O |AP_1:2,AP_2:3|
Ligand order
Ligand order information can be exported to extended CXSMILES/CXSMARTS after "LO" followed by a colon character.
-
First the center atom's index is exported followed by a colon
-
After that all atom's indexes connected to the central atom is written in ligand order separated by "."
-
The different ligand order informations are separated by comma.
-
e.g: LO:centerIdx1:idx1.idx2.idx3,centerIdx2:idx1.idx2...
Example
Cl[*](Br)I |$;_R1;;$,RG:_R1={*CCCC(C*)CC* |$_AP3;;;;;;_AP2;;;_AP1$|},{*CCCN(C*)CC* |$_AP3;;;;;;_AP2;;;_AP1$|},LO:1:0.2.3|
MDL Query features
Ring bond count (rb), Substitution count (s) and unstaturated atom (u) are exported in the following form:
rb:atomIndex1:value,atomIndex2:value
s:atomIndex1:value,atomIndex2:value
u:atomIndex1,atomIndex2,atomIndex3
Examples:
rb:1:2,2:*,4:2
u:3,4,5
Escaping
In some places special characters are escaped to '&#code' where code is the ASCII code of the special character.
Not escaped characters in fields of Sgroups and DataSgroups: 'a'-'z', 'A'-'Z', '0'-'9' and '><\"!@#$%()[]./\\?-+*^_~=' and the space character.
Not escaped characters in atom property keys and values: 'a'-'z', 'A'-'Z', '0'-'9' and '><\"!@#$%()[]./\\?-+*^_~=' and the space character.
Not escaped characters in atom labels and atom values: 'a'-'z', 'A'-'Z', '0'-'9' and '><\"!@#%()[]./\\?-+*^_~=,:' and the space character.