Huckel Analysis Plugin
This manual gives you a walk-through on how to use the Hückel Analysis Plugin:
Introduction
The Hückel Analysis Plugin is able to calculate different structural descriptors for aromatic atoms.
Localization energies L(+) and L(-) for electrophilic and nucleophilic attack at an aromatic center are calculated by the Hückel method. A smaller L(+) or L(-) value means more reactive atomic location. Order of atoms in E(+) or in Nu(-) attack are adjusted according to their localization energies. The total π energy, the π electron density and the total electron density are also calculated by Hückel method. Depending on the chemical environment, the following atoms have optimal Coulomb and resonance integral parameters: B, C, N, O, S, F, Cl, Br, I.
All other atoms have a default, non-optimized parameter. Theoretical background is taken from Isaacs' book. Additional literature for the Hückel's parameters is Streitwieser's book.
The results appear in a new window, indicating the calculated values at the atoms in the aromatic ring. In the following example the first picture is the result of Aromatic E(+)/Nu(-) order calculation, while the second picture is the result of a Pi energy calculation.
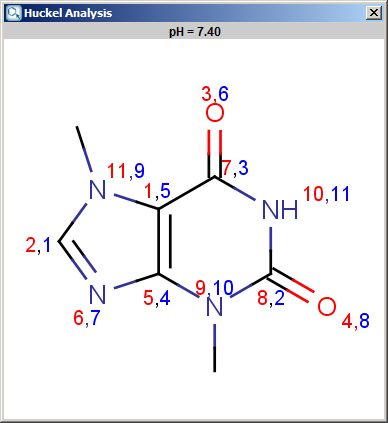
Fig. 1 Aromatic E(+)/Nu(-) calculation result
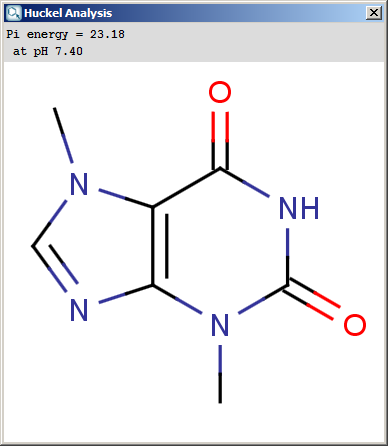
Fig. 2 Pi energy calculation result
Options
The following calculation parameters can be set in the Huckel Analysis Options window:
-
Decimal places: setting the number of decimal places with which the result value is given.
-
Type: sets the method of calculation. That could be:
-
HMO E(+)/Nu(-) order: numbers the aromatic atoms according to their probability of being attacked by electrophiles or nucleophiles.
-
HMO Localization energy L(+)/L(-): gives the localization energies of the aromatic center (dimension β).
-
HMO Pi energy: calculates the π energy of the aromatic ring(s) (dimension β).
-
HMO Electron density: calculates the π electron density.
-
HMO Charge density: calculates total charge density on the ring atoms.
-
Localization energy L(+)/L(-): the same as the HMO Localization energy L(+)/L(-), but based on experimental (reaction) data
-
Electron density: the same as the HMO Electron density, but based on experimental (reaction) data
-
Charge density: the same as the HMO Charge Density, but based on experimental (reaction) data
-
-
Subtype: E(+) or Nu(-): for E(+)/Nu(-) order and localization energy L(+)/L(-), the electrophilicity and nucleophilicity approaches can be selected (at least one of them). Results for E(+) are coloured red, and Nu(-) blue.
-
Take major microspecies at pH: calculates the values for the major microspecies at the given pH.
On the HMO method
The HMO is an abbreviation for the Hückel Molecular Orbital theory/method. The HMO calculation methods mentioned above (e.g. HMO Pi energy) uses α and β parameters based on literature data.
The non-HMO methods were developed for predicting reaction products in ChemAxon's Reactor. The α and β parameters used in these methods were determined to give the closest results to the experimental results during reaction product predictions in Reactor.
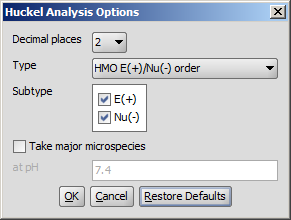
Fig. 3 Huckel Analysis Options window
References
-
Isaacs, N.S., Physical Organic Chemistry, John Wiley & Sons, Inc., New York, 1987, ISBN 0582218632.
-
Streitwieser, A., Molecular Orbital Theory for Organic Chemists, John Wiley, 1961, ISBN 0471833584.