Structure to Name User's Guide
Since version 4.1.7, Marvin contains a name generator for the evaluation of the IUPAC name or traditional name of any compound.
When possible, the generated name conforms to the Nomenclature of Organic Chemistry : IUPAC Recommendations and Preferred Names published in 2013. Our goal is to generate the preferred names specified by IUPAC in as many cases as possible, but we do not claim full conformance with that document (see below for a list of limitations). In other cases, the generated names should be systematic names that can be interpreted back to the correct chemical structure.
Importing IUPAC names is also available from version 5.1.
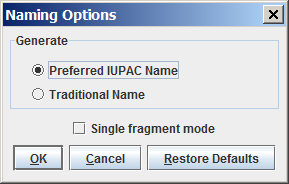
You can generate either the "Traditional Name" or the "Preferred IUPAC Name" of the molecules; you can change between these options in theNaming Options panel. By default, the "Preferred IUPAC Name" option is set. If the traditional name is requested but cannot be generated, the preferred IUPAC Name will be generated instead.
By default, molecules are handled separately if more than one molecule are drawn in the sketcher. However, sometimes a single molecule consists of more fragments (e.g., salt molecules), where the fragments should be treated as one molecule. This can be reached by switching off the "Single fragment mode" option in the Naming Options panel.
The snapshot below shows a molecule taken from the IUPAC specification, with its name computed by Marvin.
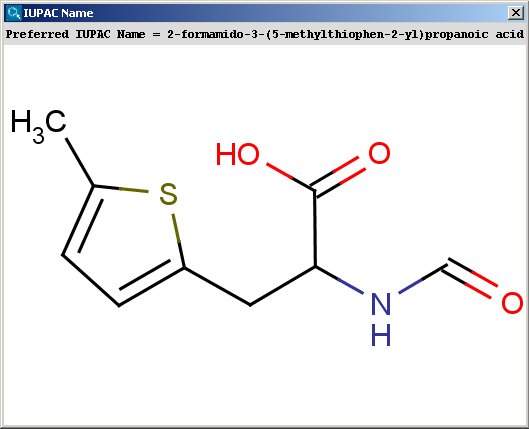
The contents of the text field can be copied to the clipboard by Ctrl+C, the structure field offers a context menu when right-clicking on it.
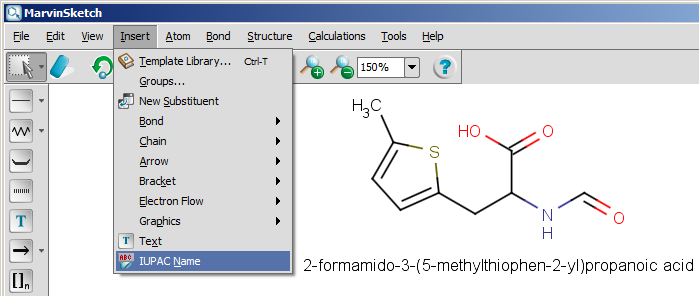
The next snapshot below shows a functionality that is available from version 5.0: the IUPAC name can be inserted into the sketch, and it changes with the structure dynamically. This functionality is available from the Structure menu by selecting the Structure to Name > Place IUPAC Name option.
Features
Supported nomenclatures include:
-
Chains, Monocycles
-
Retained/traditional names for ring systems with and without heteroatoms
-
Spiro ring systems
-
All cases of von Baeyer nomenclature for bridged ring systems
-
Ring assemblies of size 2 (e.g. biphenyl, bifuran, ...)
-
Fused ring systems (linear fused ring systems are named using the fused nomenclature, others using von Baeyer nomenclature)
-
Ethers
-
Common characteristic groups
-
Ionic compounds
-
Compounds with one radical
-
Unlimited number of atoms and rings
-
All atom types
-
Substitutive nomenclature
-
Isotopes
-
Stereochemistry
Current limitations
-
Molecules containing multiple radicals (e.g. ethane-1,2-diyl) are not supported yet.
-
Amino-acids and peptides are supported only when the amino-acids are represented as groups.
-
Molecules containing coordinate bond are not supported.
-
Some aspects of nomenclature are only partially implemented, in particular complex cases of fused systems, ring assemblies of size 3+ and multiplicative nomenclature. In those cases, a non-preferred but chemically correct name will be generated.
Usage
Individual molecules
You can name molecules by using the Naming menu entry of Tools menu in MarvinView , or Structure > Structure to Name > Generate Name in MarvinSketch .
In MarvinSketch, the name can be added to the canvas by using the Structure to Name > Place IUPAC Name entry in the Structure menu. The name will be displayed below the molecule, and updated in real-time when the molecule is modified.
Batch naming
Naming of a large number of molecules contained in a file can be achieved in two ways: with MarvinView , and on the command line, with molconvert . In both cases, all formats supported by Marvin are acceptable as input.
With MarvinView, open the file containing the structures to be names. Then select the menu File/Save As, and choose "IUPAC Name files" in the "Files of type" drop-down box. Choose a name for the file, and click on the Save button. The file will contain the names of the structures, one per line.
Alternatively, on the command line, you can use the following command:
molconvert name inputs.mol -o names.txt
The file names.txt will contain the names of the molecules in the input file, with one name per line.
It is possible to use a format option to chose a nomenclature style:
-
i (default) uses the IUPAC rules for preferred names;
-
t uses a more traditional style.
For instance, to generate traditional names, use the following:
molconvert name:t inputs.mol -o names.txt
Generate all common names for a structure:
molconvert "name:common,all" -s tylenol
Generate the most popular common name for a structure (It fails if none is known.):
molconvert name:common -s viagra
Adding names as an additional field to a SDfile can be achieved with the cxcalc tool.
cxcalc -S name input.sdf -o named.sdf
API
For information about how names can be generated from Java programs, see the developer documentation.