Screen3D
This page gives you an overview on how to use Screen3D, ChemAxon's tool for 3D virtual screening:
Introduction
Virtual Screening (VS) has become an important field in modern drug-discovery, which has mainly arisen from the increased pressure of reducing the cost of lead discovery. Currently there are two approaches to VS: ligand-based and the structure-based (docking) methods. Structure-based VS methods rely on the structural similarity between the target and the query molecule. Due to the fact that 2D fingerprints can be generated in a fast and easy way, structure-based VS methods are usually based on 2D similarity calculations. However, 2D approaches often have limitations, such as the lack of real structural insight. Therefore, 3D VS methods have been developed that also provide molecular alignments, helping e.g. pharmacophore analysis in the process of lead optimization. These methods use 3D structural similarity of the query and target pair to perform the screening.
Screen3D is a command line tool for fast 3D ligand-based virtual screening.
General information
To calculate 3D similarity for the query and target pair, Screen3D uses the flexible 3D Alignment module. The input molecule need not to have 3D coordinates; if 2D input structures are used, initial 3D conformers are automatically generated. Both molecules can be treated conformationally either rigid or flexible. In the first case the original conformation is kept during the alignment process. In the second case, the dihedral bonds of the molecule are able to freely rotate, so the conformation of the molecule changes during the superposition step.
Screen3D can only handle single-fragment molecules. Multi-fragment molecules are excluded from the input.
The result of the similarity calculation is the aligned pair of the query and the target, and the their similarity score. This is then used for ranking.
There are two types of similarity calculations available in Screen3D:
-
Shape method: this method superimposes molecules based on their 3D shape. Molecular shapes are represented by mathematical functions and the shape is coloured by attributed atom types. Atom typization can be based on ChemAxon's Dreiding force field or on pharmacophore typization.
-
Match method: this method is based on the systematic matching of atoms to be aligned.
Both methods work in two phases:
-
Input data generation: this phase "prepares" the query and target molecules for the alignment process. It calculates the shape functions, performs the necessary atom typization etc. The output of this phase are two files: a binary file used in the second phase and a molecule file to view the 3D structure.
-
Screening phase: this phrase performs the actual screening. The output of this phase is different similarity-related data (unnormalized and Tanimoto similarity) in a text format and a structure file with the aligned target structures.
Details on how these methods work (and when and how they are applicable) can be found in the reference article.
Usage example
The following simple workflow shows how to use the Screen3D command line tool. All options of the command line tool can be seen by running
screen3d -h
Simple screening workflow with Screen3D
We implement the following screening workflow with Screen3D: we want to find a molecule that will possibly mimic the biological activity of a known drug. The drug sturcture is available as a co-crystal ligand structure in an MRV file. The target structures are only available as SMILES codes in a separate file.
In this case the best choice is to keep the original ligand structure intact by setting it rigid during the alignment. However, as we don't have fixed conformers for the target structures, we have to keep them flexible during the alignment process.
So the first (pre-processing) phase for the ligand and target structures is, respectively:
screen3d g ligand.mrv screen3d g targets.smiles -F In this case we use the Shape method with extended atom types for the alignment (default setting). The output are two .ser (binary) files that contain the pre-processed structures.
The second (screening) phase we use the following command:
screen3d s -t targetsF.ser -q ligandF.ser -conf 4 -oformat sdf -c "Screening"The options used set the number of generated conformers for the non-rigid structures. The output format will be SDF, and we also print the simple comment "Screening".
The first few lines of the output of this command give information on the screening process. After them come the screening results written to the standard output:
Similarity calculation is started
Query file : ligandF.ser
Target file: targetsF.ser
Will write aligned structures to: ligandF_targetsF_shape_1418652211953_aligned.sdf
seq Unnormalized3DScore 3DTanimoto t(ms) MW(target) n_rot_bonds(target)
0 97,88 0,16 340 115,99 0 0
1 283,12 0,31 1597 323,89 3 0
2 261,89 0,43 380 214,95 0 0
3 30,37 0,05 130 141,97 1 0
4 282,06 0,35 830 213,99 0 0
5 294,10 0,27 1540 477,81 2 0
6 356,05 0,60 748 224,96 1 0
7 314,29 0,38 667 257,98 0 0
8 61,84 0,10 150 108,00 1 0
...
This text output is also printed into a .txt file which has the same prefix name as the structural output file.
You can create an enrichment graph for your screening process. To do this, you have to run the screening for your set of active and inactive molecules. After this run the command
screen3d e inactives_output.txt actives_output.txt
Screen3D in KNIME
3D virtual screening is also available in the KNIME workflow management system. This system allows to assemble workflows for different tasks. A KNIME workflow is basically a chain of nodes, where each node performs a specific task. The screening workflow above can be implemented in KNIME by the following chain of nodes:
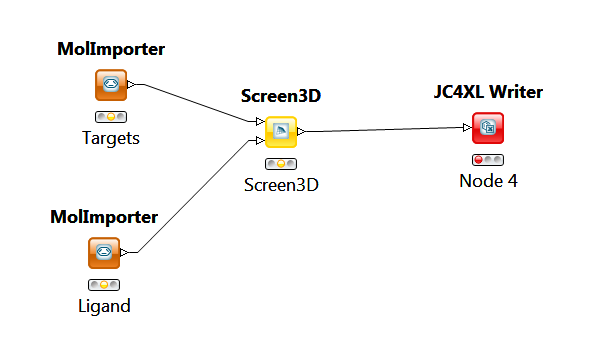
The virtual screening is performed by the Screen3D node. This node has two input nodes and one output node. In this case the two input nodes are two MolImporter nodes that can read molecules from a specifed file. The output node is a JC4XL node that writes the output into an Excel file.
You can get a full description of the node by single-clicking on it in KNIME. The node description appears in a separate window on the right side of the KNIME window.
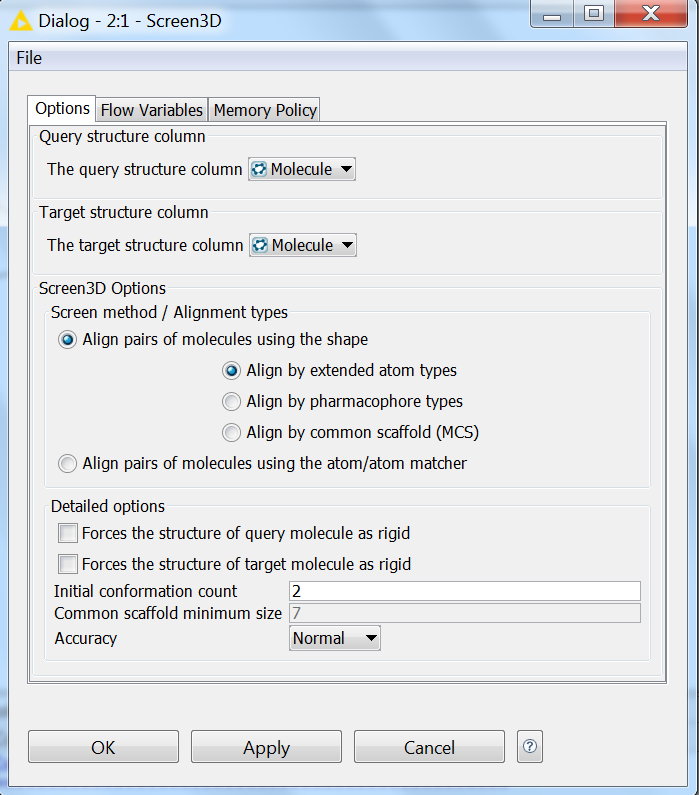
All options that are available in the Screen3D command line tool can also be reached via the Screen3D KNIME node. To set the screening options use Right Click > Configure on the Screen3D node. With this the following dialog window pops up:
References
-
Kalászi, A.; Szisz D.; Imre G.; Polgár T., Screen3D: A Novel Fully Flexible High-Throughput Shape-Similarity Search Method. J. Chem. Inf. Model. , 2014 , 54 (4), pp 1036–1049