Working examples
Contents
Introduction
These examples demonstrate the use of the Reactor program. The purpose is to show how to run the react command and explain some of its command line options.
For a description of reaction rules and mapping, see the Reaction rules and Reaction mapping sections of the Reactor Manual.
Reaction rule working examples showing more complex combinations of the reactivity, exclude, and selectivity rules are also available.
Prerequisites
These examples run the react UNIX shell script under UNIX / Linux or the react.bat batch file under Windows.
To run these examples:
-
The Java Virtual Machine version 1.4 or higher and JChem have to be installed on your system.
-
The PATH environment variable has to be set as described in the Preparing and Running JChem's Batch Files and Shell Scripts manual.
-
A command shell (under UNIX / Linux: your favorite shell, under Windows: a Cygwin shell or a Command Prompt) has to be run in the reactor example directory.
In UNIX / Linux:cd jchem/examples/reactor
In Windows:
cd jchem\examples\reactor
Simple examples
These examples demonstrate simple reaction processing with different combinations of the most popular command line options. The reaction file is reaction.rxn and the two reactants are stored in reactant1.mol and reactant2.mol.
-
For the simplest reaction processing type:
react -r reaction.rxn reactant1.mol reactant2.mol
or else you can also give the reaction and the reactants in SMARTS/SMILES string form:
react -r "[F,Cl,Br,I:3][C:1]=O.[H:4][O:2][#6]>>[#6][O:2][C:1]=O.[F,Cl,Br,I:3][H:4]" \
"FC(=O)C1=CC2=C(C=CC(CC(Br)=O)=C2)N(C(Cl)=O)C3=C1C=CC=C3" "CC(=O)OCCN1CCN(CCO)CC1"The -r command line option specifies the reaction file or string, while input files/SMILES strings (without preceeding command line option) are taken as reactants. The resulting products are written to the standard output in SMILES format:
CC(=O)OCCN1CCN(CCOC(=O)N2C3=C(C=C(CC(Br)=O)C=C3)C=C(C(F)=O)C4=C2C=CC=C4)CC1
Cl
CC(=O)OCCN1CCN(CCOC(=O)C2=CC3=C(C=CC(CC(Br)=O)=C3)N(C(Cl)=O)C4=C2C=CC=C4)CC1
F
CC(=O)OCCN1CCN(CCOC(=O)CC2=CC3=C(C=C2)N(C(Cl)=O)C4=C(C=CC=C4)C(=C3)C(F)=O)CC1
Br -
The result is written in reaction form if the -t command line option is specified with reaction output type:
react -r reaction.rxn -t reaction reactant1.mol reactant2.mol
The result in this case is:
FC(=O)C1=CC2=C(C=CC(CC(Br)=O)=C2)N(C(Cl)=O)C3=C1C=CC=C3.CC(=O)OCCN1CCN(CCO)CC1>>CC(=O)OCCN1CCN(CCOC(=O)N2C3=C(C=C(CC(Br)=O)C=C3)C=C(C(F)=O)C4=C2C=CC=C4)CC1.Cl
FC(=O)C1=CC2=C(C=CC(CC(Br)=O)=C2)N(C(Cl)=O)C3=C1C=CC=C3.CC(=O)OCCN1CCN(CCO)CC1>>CC(=O)OCCN1CCN(CCOC(=O)C2=CC3=C(C=CC(CC(Br)=O)=C3)N(C(Cl)=O)C4=C2C=CC=C4)CC1.F
FC(=O)C1=CC2=C(C=CC(CC(Br)=O)=C2)N(C(Cl)=O)C3=C1C=CC=C3.CC(=O)OCCN1CCN(CCO)CC1>>CC(=O)OCCN1CCN(CCOC(=O)CC2=CC3=C(C=C2)N(C(Cl)=O)C4=C(C=CC=C4)C(=C3)C(F)=O)CC1.Br -
You can also save the result in a file by specifying the output file in the -o option:
react -r reaction.rxn -t reaction -o result.smiles reactant1.mol reactant2.mol
-
A different output file format can be specified in the -f option:
react -r reaction.rxn -t reaction -f rxn -o result.rxn reactant1.mol reactant2.mol
-
Now go back to the first example. You may want to get rid of the second products. You can do this by specifying the required product index in the -x option:
react -r reaction.rxn -x 1 reactant1.mol reactant2.mol
The result is:
CC(=O)OCCN1CCN(CCOC(=O)N2C3=C(C=C(CC(Br)=O)C=C3)C=C(C(F)=O)C4=C2C=CC=C4)CC1
CC(=O)OCCN1CCN(CCOC(=O)C2=CC3=C(C=CC(CC(Br)=O)=C3)N(C(Cl)=O)C4=C2C=CC=C4)CC1
CC(=O)OCCN1CCN(CCOC(=O)CC2=CC3=C(C=C2)N(C(Cl)=O)C4=C(C=CC=C4)C(=C3)C(F)=O)CC1Note, that multiple product indexes can also be specified by an index list separated by commas, without spaces (e.g. select the first and the third from a 3-product reaction by: -x 1,3).
-
Save the previous output in SDF format:
react -r reaction.rxn -x 1 -f sdf -o result.sdf reactant1.mol reactant2.mol
Show the result in MView:
mview result.sdf
Note, that under UNIX/Linux it is also possible to pipe the result directly to MView without saving it:
react -r reaction.rxn -x 1 reactant1.mol reactant2.mol | mview -
-
Go back to the first example and save the first product list (specify the number of results to be returned in -p):
react -r reaction.rxn reactant1.mol reactant2.mol -p 1 -o products.smiles
Assume that you have products.smiles and you want to know the reactants that can be formed to these products by processing the reaction reaction.rxn. That is, you want to process the reaction in reversed direction. You can do this by the -s option:
react -r reaction.rxn products.smiles -s
The result is:
CC(Cl)=O
OCCN1CCN(CCOC(=O)N2C3=C(C=C(CC(Br)=O)C=C3)C=C(C(F)=O)C4=C2C=CC=C4)CC1
FC(=O)C1=CC2=C(C=CC(CC(Br)=O)=C2)N(C(Cl)=O)C3=C1C=CC=C3
CC(=O)OCCN1CCN(CCO)CC1which includes our two original reactants.
The use and meaning of command-line options in the above commands:
|
Option |
Description |
Default |
|
-r |
specifies the reaction file |
- |
|
-t |
specifies the output type ('reaction' or 'product') |
'product' |
|
-f |
specifies the output format (e.g. 'sdf', 'mol', 'rxn') |
'smiles' |
|
-o |
specifies the output file path |
standard output (console) |
|
-x |
extract specified products (e.g. 1,3) |
return all products |
|
-p |
the maximum number of product lists to be returned (e.g. 2) |
return all product lists |
|
-s |
take reversed reaction |
take reaction as it is |
Multiple reactants
These examples demonstrate reaction processing on multiple reactants: a molecule file with possibly more than one molecule structures is specified in place of each reaction reactant. Reactor can process multiple reactants in two modes, sequential or combinatorial (default: sequential). In the examples below, the first reactant filereactant1.smiles contains 2 molecules and the second reactant file reactant2.smiles contains 3 molecules.
-
Process the reaction in sequential mode (default) with reaction output type:
react -r reaction.rxn reactant1.smiles reactant2.smiles -t reaction
The result is:
ClC(=O)N1C2=CC=CC=C2C=CC3=C1C=CC=C3.CCC1=CC=C(O)C=C1>>CCC1=CC=C(OC(=O)N2C3=CC=CC=C3C=CC4=C2C=CC=C4)C=C1.Cl
CCC1=CC=C(CC(Cl)=O)C=C1.CCCO>>CCCOC(=O)CC1=CC=C(CC)C=C1.Cl
CCC1=CC=C(CC(Cl)=O)C=C1.OC1CCCCC1>>CCC1=CC=C(CC(=O)OC2CCCCC2)C=C1.ClNote, that the last (second) molecule in the first reactant file reactant1.smiles is repeatedly paired with both the second and the third molecule in the second reactant filereactant2.smiles since the first reactant file contains less molecules than the second reactant file.
-
Process the reaction in combinatorial mode with reaction output type:
react -r reaction.rxn reactant1.smiles reactant2.smiles -m comb -t reaction
The result is:
ClC(=O)N1C2=CC=CC=C2C=CC3=C1C=CC=C3.CCC1=CC=C(O)C=C1>>CCC1=CC=C(OC(=O)N2C3=CC=CC=C3C=CC4=C2C=CC=C4)C=C1.Cl
ClC(=O)N1C2=CC=CC=C2C=CC3=C1C=CC=C3.CCCO>>CCCOC(=O)N1C2=CC=CC=C2C=CC3=C1C=CC=C3.Cl
ClC(=O)N1C2=CC=CC=C2C=CC3=C1C=CC=C3.OC1CCCCC1>>O=C(OC1CCCCC1)N2C3=CC=CC=C3C=CC4=C2C=CC=C4.Cl
CCC1=CC=C(CC(Cl)=O)C=C1.CCC1=CC=C(O)C=C1>>CCC1=CC=C(CC(=O)OC2=CC=C(CC)C=C2)C=C1.Cl
CCC1=CC=C(CC(Cl)=O)C=C1.CCCO>>CCCOC(=O)CC1=CC=C(CC)C=C1.Cl
CCC1=CC=C(CC(Cl)=O)C=C1.OC1CCCCC1>>CCC1=CC=C(CC(=O)OC2CCCCC2)C=C1.ClNote, that there are 2*3=6 reactions, since all reactant pairs are processed with the first reactant taken from reactant1.smiles and the second from reactant2.smiles.
The use and meaning of command-line options in the above commands:
|
Option |
Description |
Default |
|
-m |
specifies the processing mode |
'seq' |
Reactivity rules
These examples show how to specify a reactivity rule on reactants and/or products: reaction products accepted only if the reactivity rule is satisfied. Rules are defined aschemical terms and evaluated by the Chemical Terms Evaluator. See the Reaction rules section of the Reactor Manual for the syntax of these rules.
-
The reactivity rule in this example is defined in an RDF tag of the reaction file and describes amine-type nitrogens: a N atom is taken as an amine-type nitrogen if and only if it is either an amine or a hydrazine(N2) but not a hydrazine(N1) and not an amide.
hydrazine
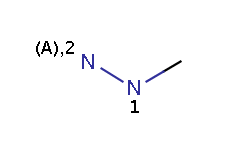
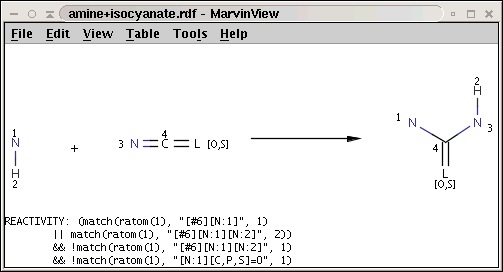
The reaction file amine+isocyanate.rdf contains the reactivity rule in the REACTIVITY RDF tag (you can see it in MView by setting Table / Show Fields):
Our reactant files are amines.smiles and isocyanates.smiles:
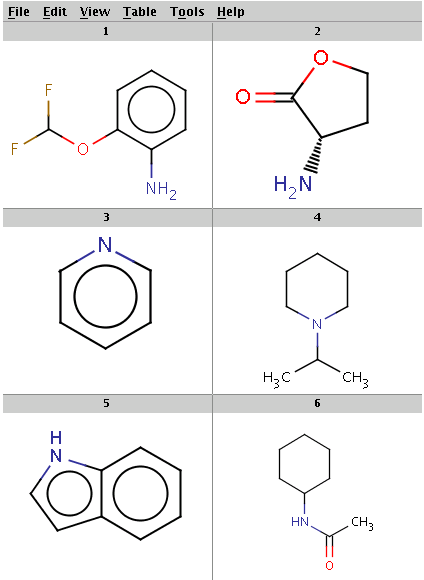
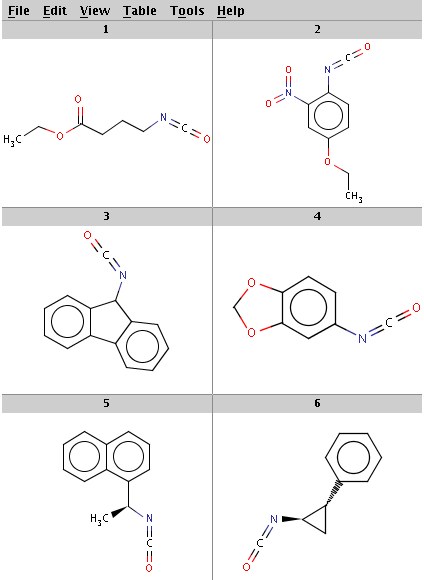
Look at the amines: The first and the second amine satisfy the condition, the next two are not attached to a hydrogen therefore do not satisfy the reaction equation itself, the remaining two do not satisfy the reactivity rule above. In the examples below, Reactor is run with multiple reactants in sequential mode: the first amine is paired with the first isocyanate, then the second amine with the second isocyanate, and so on.
Run Reactor by:
react -r amine+isocyanate.rdf amines.smiles isocyanates.smiles -t reaction
The result is:
Nc1ccccc1OC(F)F.CCOC(=O)CCCN=C=O>>CCOC(=O)CCCNC(=O)Nc1ccccc1OC(F)F
N[C@H]1CCOC1=O.CCOc1ccc(N=C=O)c(c1)N(=O)=O>>CCOc1ccc(NC(=O)N[C@H]2CCOC2=O)c(c1)N(=O)=OYou can see that only the first two amines have been processed:
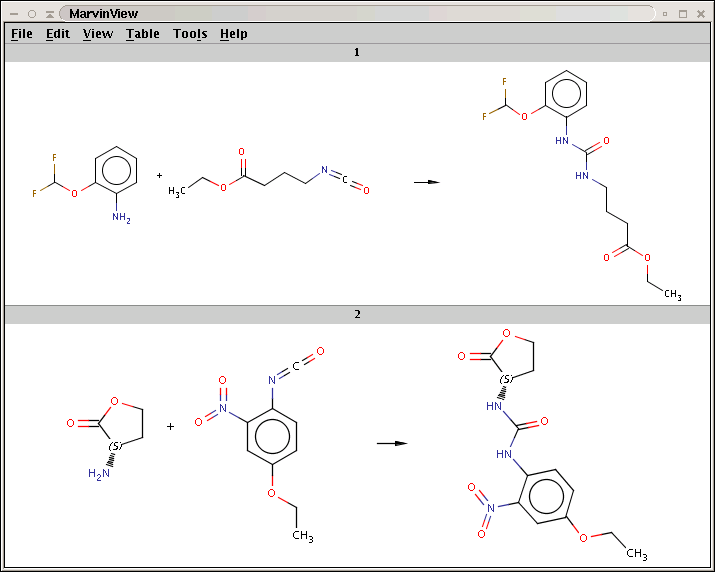
For comparison, run Reactor without reaction rules (option -n):
react -r amine+isocyanate.rdf amines.smiles isocyanates.smiles -t reaction -n r
We have 4 result rows corresponding to the first two and the last two amines, since the reaction equation is not satisfied for the third and the fourth amines:
Nc1ccccc1OC(F)F.CCOC(=O)CCCN=C=O>>CCOC(=O)CCCNC(=O)Nc1ccccc1OC(F)F
N[C@H]1CCOC1=O.CCOc1ccc(N=C=O)c(c1)N(=O)=O>>CCOc1ccc(NC(=O)N[C@H]2CCOC2=O)c(c1)N(=O)=O
c1ccc2[nH]ccc2c1.C[C@H](N=C=O)c1cccc2ccccc12>>C[C@H](NC(=O)[nH]1ccc2ccccc12)c3cccc4ccccc34
CC(=O)NC1CCCCC1.O=C=N[C@@H]1C[C@H]1c2ccccc2>>CC(=O)N(C1CCCCC1)C(=O)N[C@@H]2C[C@H]2c3ccccc3 -
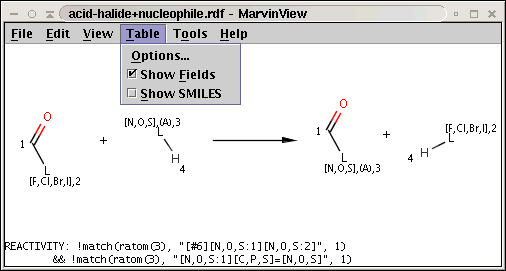
In our next example the reaction is stored in acid-halide+nucleophile.rdf and the corresponding conditionial rule is applied to nucleophiles, the second reactant of the reaction:
!match(ratom(3), "[#6][N,O,S:1][N,O,S:2]", 1) && !match(ratom(3), "[N,O,S:1][C,P,S]=[N,O,S]", 1)
The reaction with the rule is shown below:
The two matching conditions say that the reaction center of the nucleophile (reactant atom with map 3) should not match the atom with map 1 in any of the following molecule structures:
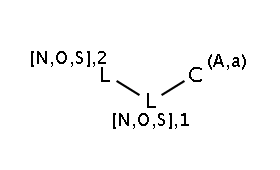
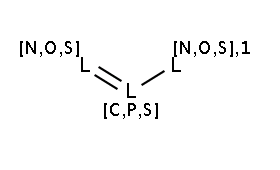
The reactant files are acid-halides.smiles and nucleophiles.smiles. Four sample molecules from each are shown below:
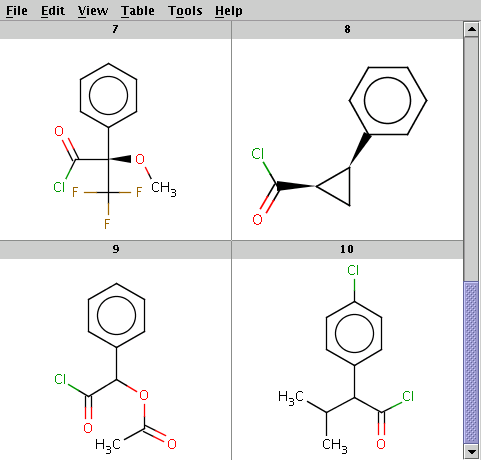
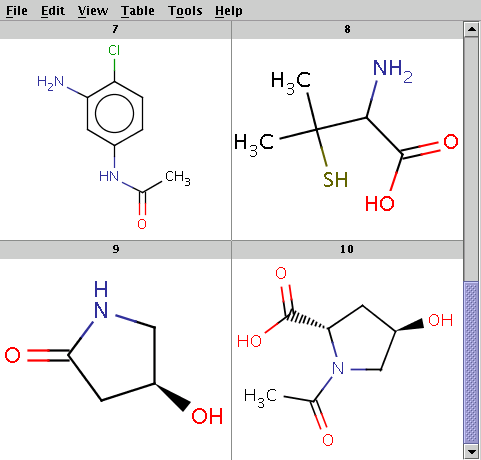
Run Reactor in combinatorial mode (-m comb), extract the first product (-x 1):
react -r acid-halide+nucleophile.rdf acid-halides.smiles nucleophiles.smiles -m comb -x 1 -o acid-halide+nucleophile_result.smiles
The result is stored in acid-halide+nucleophile_result.smiles. Some sample products are shown below:
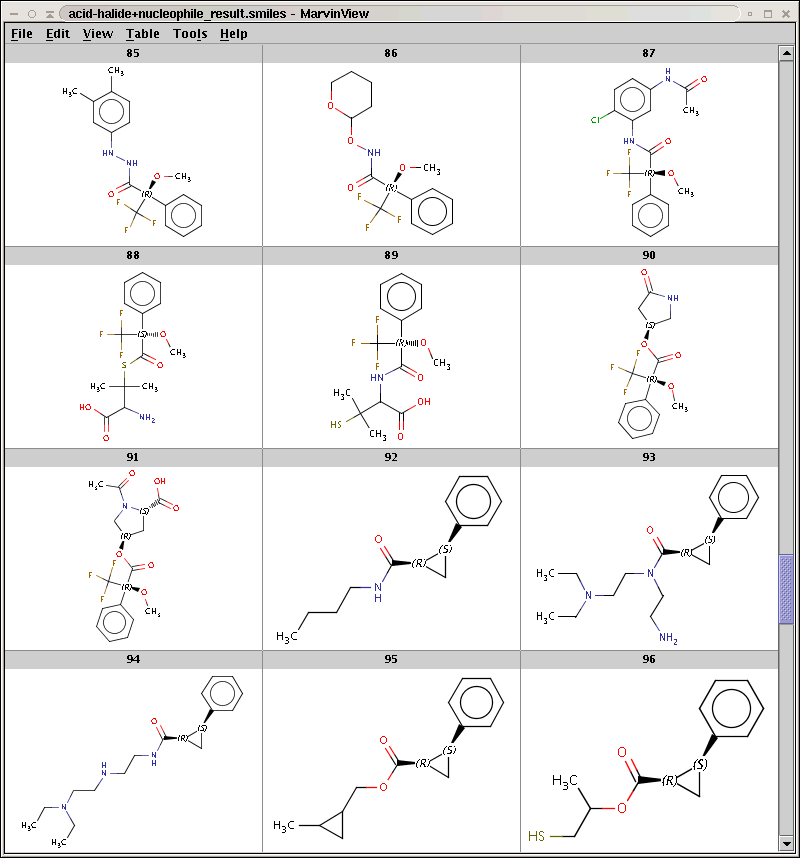
By default, Reactor filters product repetitions resulting from processing symmetric reaction centers. To increase efficiency, it is sometimes useful to allow product repetitions by skipping this duplicate check with the -w option. Run Reactor with this option:
react -r acid-halide+nucleophile.rdf acid-halides.smiles nucleophiles.smiles -m comb -x 1 -w -o acid-halide+nucleophile_result_dup.smiles
Observe, that the result file with duplicates acid-halide+nucleophile_result_dup.smiles contains 190 structures, while the result file without duplicates acid-halide+nucleophile_result.smiles contains only 130 structures.
Now run Reactor without reaction rules (option -n):
react -r acid-halide+nucleophile.rdf acid-halides.smiles nucleophiles.smiles -m comb -x 1 -n r -o acid-halide+nucleophile_result_norules.smiles
The result is stored in acid-halide+nucleophile_result_norules.smiles.
Observe, that with the application of reaction rules we have 130 products in acid-halide+nucleophile_result.smiles, while ignoring reaction rules results in 180 products in acid-halide+nucleophile_result_norules.smiles.
-
A generalization of the amine+isocyanate reaction in our first reactivity rule example can be obtained by the application of the matching rule in our second reactivity rule example:
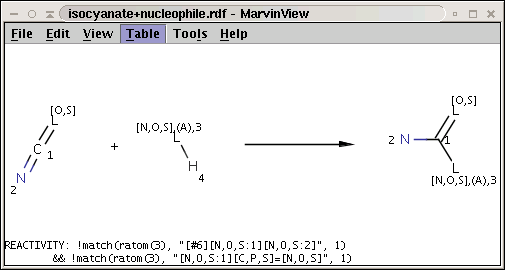
The reaction file is isocyanate+nucleophile.rdf. The reactant files are isocyanates_more.smiles and nucleophiles.smiles. Four sample molecules from each are shown below:
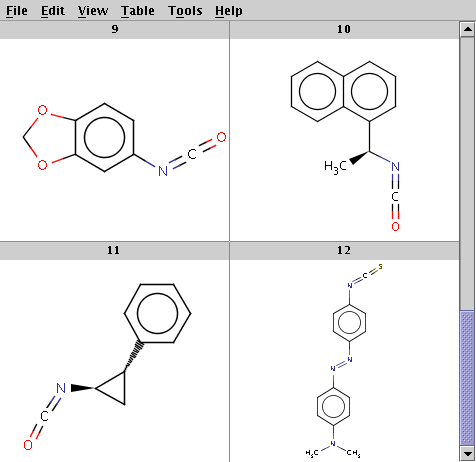
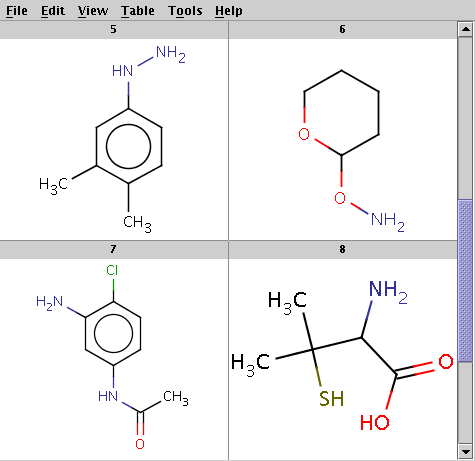
Run Reactor in combinatorial mode (-m comb):
react -r isocyanate+nucleophile.rdf isocyanates_more.smiles nucleophiles.smiles -m comb -t reaction -o isocyanate+nucleophile_result.smiles
The result is stored in isocyanate+nucleophile_result.smiles. The two sample results below show that the same reactants can be transformed to different products by choosing different reaction centers:
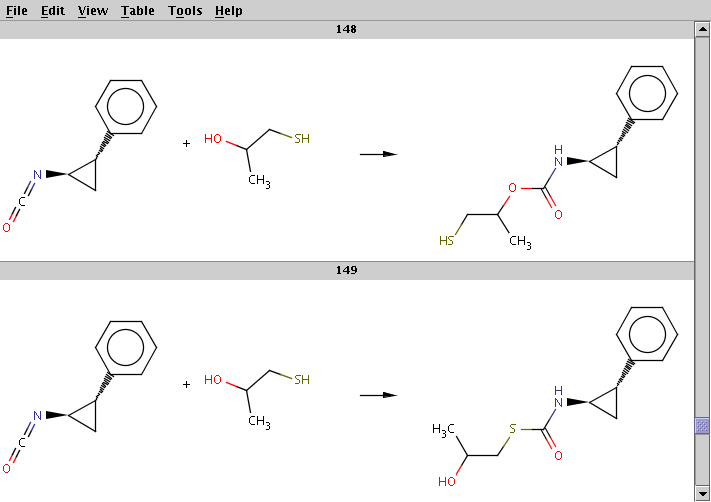
For comparison, run Reactor without reaction rules (option -n):
react -r isocyanate+nucleophile.rdf isocyanates_more.smiles nucleophiles.smiles -m comb -t reaction -n r -o isocyanate+nucleophile_result_norules.smiles
Compare the number of results: with the application of the rules we have 169 results, while without the rules we have 234 results.
Note, that reactions with rules can be defined either in an RDF/MRV file with the rule specified in an RDF/MRV tag as shown above, or else in the reaction string as shown in the Selectivity rule examples below.
The use and meaning of command-line options in the above commands:
|
Option |
Description |
Default |
|
-n |
ignore reaction rules |
process reaction rules |
|
-w |
allow duplicate product lists |
filter product repetitions |
Selectivity rules
These examples show how to specify a selectivity rule on reactants and/or products: reaction products are always accepted but sorted according to priority defined by reaction rules. Rules are defined as chemical terms and evaluated by the Chemical Terms Evaluator. See the Reaction rules section of the Reactor Manual for the syntax of these rules.
Note, that the Tolerance attribute sets the sensitivity of the selectivity maximum: product lists with selectivity value falling within tolerance of the maximum possible selectivity value are accepted, others rejected. The default tolerance is 0.0001, which means that real-valued selectivity values should be approximately equal to the maximum, while integral selectivity values are tested for equality with the maximum. If you want to accept all product lists and use the selectivity rule only for sorting the results then apply the -n t command line option in which case Reactor ignores the tolerance setting.
In RDF files, the selectivity rule can be set in the SELECTIVITY RDF tag, while the tolerance can be specified in a separate TOLERANCE RDF tag.
-
This example demonstrates the rule that bromine is attracted to the aromatic carbon atom having the lowest localization energy having an attached electrophile in the intermediate state.
To run this example, license for the HuckelAnalysisPlugin is required (otherwise Reactor can calculate the energy values for the first reactant only).
The reaction can be described with the following SMARTS string:
[c:1][H:2]>>[c:1][Br:3]
while the selectivity rule can be defined with this Chemical Terms expression:
-energyE(ratom(1))
These together define the following reaction:
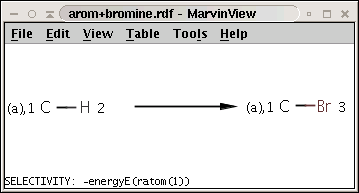
Note the minus sign in front of the rule: this means that we want to sort results in increasing order instead of decreasing order, that is, we prefer the aromatic carbon (attached to a hydrogen) with lowest localization energy having an attached electrophile in the intermediate state.
Note, that this simple rule is applicable for benzene derivatives only.
Our sample reactants are stored in the arom.smiles molecule file:
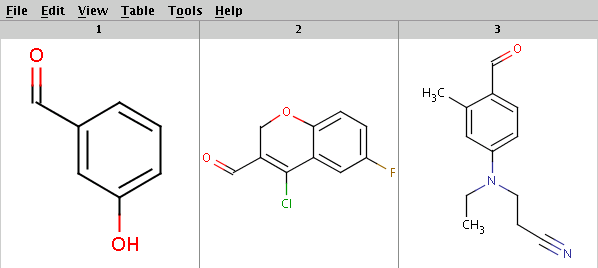
Run Reactor with default tolerance to get the products with highest priority (determined by the evaluation result of the selectivity rule):
react -r "[c:1][H:2]>>[c:1][Br:3]..s:-energyE(ratom(1))" arom.smiles
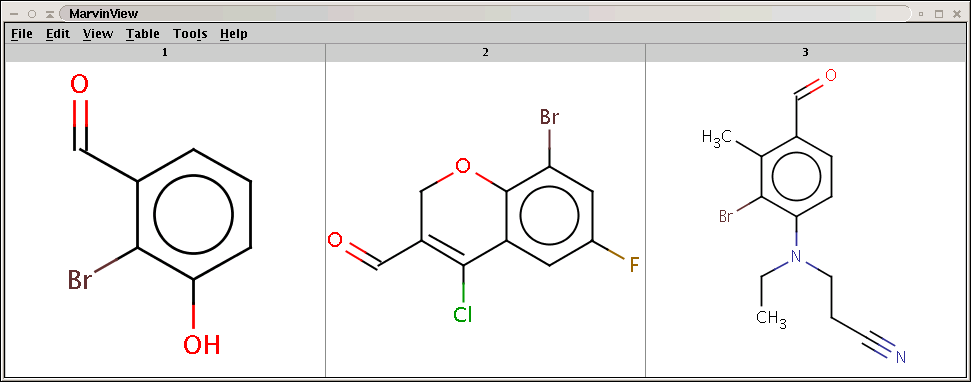
The result is:
OC1=CC=CC(C=O)=C1Br
FC1=CC(Br)=C2OCC(C=O)=C(Cl)C2=C1
CCN(CCC#N)C1=CC=C(C=O)C(C)=C1BrCheck that the bromine indeed became attached to the aromatic carbon (with hydrogen) with lowest localization energy having an attached electrophile in the intermediate state:
Reactants with electrophilic localization energy values
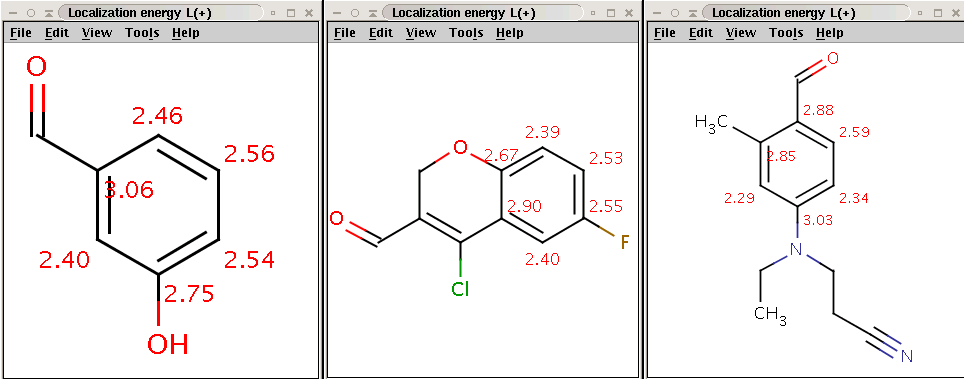
Products with bromine attached to the aromatic carbon (with hydrogen) having lowest electrophilic localization energy
Standardization of reactants can be defined in the STANDARDIZATION RDF tag of the reaction molecule or after the -S command line option. The standardization specified using the command line option overrides the standardization given in the STANDARDIZATION RDF tag. For the description of standardization and its configuration, refer to the Standardizer Manual.
Running the same example with aromatization specified after the -S option as a Standardizer action string:
react -r "[c:1][H:2]>>[c:1][Br:3]..s:-energyE(ratom(1))" -S aromatize arom.smiles
The result is now aromatized:
Oc1cccc(C=O)c1Br
Fc1cc(Br)c2OCC(C=O)=C(Cl)c2c1
CCN(CCC#N)c1ccc(C=O)c(C)c1Br -
This example demonstrates a selectivity rule with calculating the hydrogen count: when adding hydrogen chloride to an alkene, the carbon-carbon double bond is transformed to a single bond with the chloride being attached to the carbon with less hydrogens (Markovnikov rule).
The reaction (alkene+hcl.rdf) with the selectivity rule is shown below:
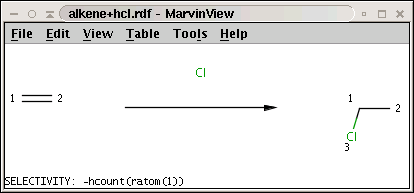
Note the minus sign in front of the rule: this means that we want to sort results in increasing order instead of decreasing order, that is, we prefer the carbon with least number of attached hydrogens.
The reactants are taken from alkenes.smiles. Four sample reactants are shown below:
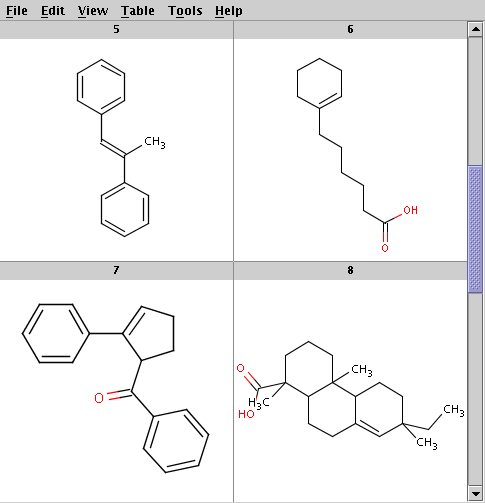
Run Reactor with the default tolerance to get the product with highest priority (determined by the evaluation result of the selectivity rule):
react -r alkene+hcl.rdf alkenes.smiles -S aromatize.xml -t reaction -o alkene+hcl_result.smiles
This time we used aromatize.xml to specify the Standardizer configuration. It only contains aromatization.
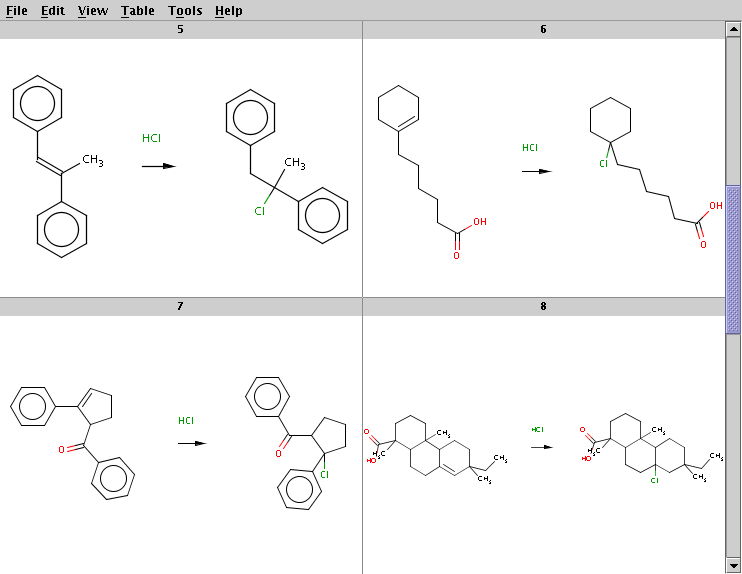
The result is stored in alkene+hcl_result.smiles. Four sample result reactions are shown below:
The Cl atom is attached to the carbon with less hydrogens
Here is another invocation of the same reaction processing, using SMARTS reaction string with appended selectivity rule:
react -r "[C:1]=[C:2]>Cl>[C:1]([#6:2])[Cl:3]..s:-hcount(ratom(1))" alkenes.smiles -S aromatize.xml -t reaction
The result is the same as above.
The use and meaning of command-line options in the above commands:
|
Option |
Description |
Default |
|
-S |
Standardize reactants |
- |
Do you have a question? Would you like to learn more?
Please browse among the related topics on our support forum or search the website. If you want to suggest modifications or improvements to our documentation email our support directly!
![]()
RESOURCES
ABOUT US
NEWSLETTER
SubscribePeople @ ChemAxon
Copyright © 1998-2015 ChemAxon Ltd. All rights reserved. EULA Privacy Policy