Administration Guide - JChem Manager
The Administration Guide of JChem provides background information about the administration of structure tables in relational databases using JChemManager. It contains information about table creation, importing and exporting structure files.
Contents
1. System requirements
In order to run JChemManager the following software need to be installed:
-
JChem for end users and Java developers (See installation guide here.)
-
Java Virtual Machine version 6 or later.
-
A relational database engine (RDBMS) that will store the structures.
Supported database systems:-
Oracle
-
MySQL
-
IBM DB2
-
Microsoft SQL Server
-
HSQLBD
-
Microsoft Access
-
Derby
-
PostgreSQL
-
Composite (Read only)
The database server doesn't have to be installed on the same machine as JChemManager if the computers are connected in a local network (Internet connection is also sufficient, but the speed of such a system might be low).
If needed, create a new database and/or a new user for the database with appropriate rights. -
-
A JDBC driver for the database engine * , which is compatible with the applied version of Java Virtual Machine. The driver has to be in the CLASSPATH environment variable on the same computer as JChemManager. JChem suite contains bundled JDBC drivers for the supported databases. If the bundled driver is not appropriate for your case, you can override it with another JDBC driver. This is usually a .jar file, which is either distributed with the database, or can be downloaded from the vendor's website. Please note, that the actual file should be listed in the CLASSPATH before the bundled driver.
* For Microsoft Access JChem uses the ODBC driver included in Java, no external driver needed. ODBC connection to other database engines is not supported.
You can find information about the hardware requirements here.
2. About JChemManager
JChemManager is a tool for creating, deleting structure tables, and importing, exporting structure files into and out of these tables. The program is a two-tier Java application that can be run under operating systems supplied with Java (see for more on the System requirements).
Stored settings:
-
JChemManager stores user-specific settings in the .jchem file located in the .chemaxon (UNIX / Linux) or chemaxon (Windows) directory in the user's home directory.
-
General options and information needed for using structure tables are stored in the JChem property table (default name: JChemProperties)
3. Running JChemManager
Starting JChemManager GUI from command line
Prepare the usage of the jcman script as described in Preparing the Usage of JChem Batch Files and Shell Scripts. Run JChemManager by entering
jcman Warning:
To avoid the conflict of different versions of classes, jchem.jar should not be included in the system's class path (CLASSPATH). See jcman or jcman.bat in the bin subdirectory as examples. Avoid using directory names with spaces (e.g. use PROGRA~1 instead of Program Files in Win32) For example, such problem might occur when you view a HTML page using a browser, which contains a Marvin applet and, at the same time, the system's class path contains jchem.jar.
Command line usage
Many operations of jcman can also be invoked without graphic interface. The command line usage makes the administration easier from a remote machine
-
where Java or JChem is not installed or
-
when the connection is slow
In the above cases connect to the server using telnet or ssh and invoke jcman. The list of options (as listed with jcman -h):
Usage of GUI program:
jcman
Usage of command line program:
jcman <command> [options]
Commands:
c <table> create table in database
t [--rowcount] list structure tables with or without row numbers
t <table> show information on a structure table
t <table> <row> show the specified row
v show version information
a <table> <file> import (add molecules) from a file into a table
x <table> <format> export table to standard output.
format:
sdf (MDL SDfile)
mol (MDL Molfile)
rdf (MDL RDMolfile)
smi (Daylight smiles)
mol2 (Tripos Mol2 file)
inchi (IUPAC InChI file)
mrv (Marvin document)
x <table> <file> export a table into a file. (The format will be
determined from the extension:.sdf/.mol/.smi
/.rdf/.mol2/.inchi/.mrv)
x <table> <file> <field>[:<field>]
export specific fields from a table into a file
x <table> <file> |<dbColumnName>:<outputName>[|<dbColumnName>:<outputName>]|
replace the original names of database columns
to user defined names in the export output file;
can be used together with the previous export option
d <table> drop (remove) a structure table
d <table> -where <"where clause">
delete all entries that satisfy the conditions
(valid 'where clause' between double quotes) from
a structure table
u upgrades database structure and recalculates all old
tables if necessary (typically after upgrade)
u <table> upgrades database structure and recalculates an old
table if necessary (typically after upgrade)
u --ptid upgrades database structure (with recalculation) and
generates a new unique identifier for the currently
used property table. This is needed by the cache if
you use multiple databases or schemas with different
property tables handling the same JChem table names.
r recalculate all structure tables except Chemical
Terms columns
r <table> recalculate a structure table except Chemical Terms
r --ct <table> recalculate only the Chemical Terms columns of a
structure table
r --md [<table>] [<desc table>]
recalculate only the Molecular Descriptors
k precalculate all structure tables without Chemical Terms
k <table> precalculate a structure table without Chemical Terms
k --remove [<table>] remove precalculated data for one or for every table
s <table> calculate and prints statistics for table
m <table> miscellaneous operations on the specified table
g <global-option> set options affecting all tables
Options (general):
-h --help this help message
--driver <JDBC driver> the JDBC driver to use
--dburl <url> the database URL to connect
--proptable <table> the name of property table
-l --login <login> login name
-p --password <password> password
-s --saveconf save settings into
" _<an actual directory in your file system appears here>_ "
Options for table creation:
--fplength <n> fingerprint size in bits
(default depends on table type)
--bits <n> bits to be set for patterns
(default depends on table type)
--bonds <n> pattern length
(default depends on table type)
--coldefs <column defs> column definitions. If not empty, then syntax is
", name1 type1, name2 type2, ..."
see doc of CREATE TABLE on how to define columns
--stconfig <file> standardizer configuration. If not given, default
standardization is used.
--relative only treats as absolute stereo if chiral flag set.
--nodup does not import molecules already in database
--ctcolcfg <cols-exprs> a semicolon separated list of pairs of
column names and Chemical Terms expressions.
Each pair specifies that the values of the given
column should be automatically calculated using
the given Chemical Terms expression.
In each pair, the column name and the
Chemical Terms expression is separated with
an equal sign ('=').
--t:<type> the type of the structure table
--t:molecules Specific structures, like single molecules,
mixtures, salts, polymers
--t:any All types of structures are allowed, but no
structure type-specific searching
--t:reactions single step reactions
--t:markush for the storage of Markush structures (this table
type is not allowed for MS Access DBMS)
--t:query query structures
--tds:[y/n] specify "y" to consider tautomers during duplicate
search (default is "n")
Option for recalculation :
--stconfig <file> standardizer configuration. If not given, there is
no change in the standardizer configuration.
Specify "reset_to_default" to change to default
standardization.
--tds:[y/n] specify "y"to consider tautomers during duplicate
search (default is "n")
Options for import:
--connect <connections> assign custom table fields to SDFile tags
Note: If not given, only those SDFile tags will be
imported that have identical field names in the
structure table
Example: --connect "dbfield1=sdfield1;dbfield2=sdfield2"
--nametofield <field> name of the database field that should store the
structures' names
--skip <n> skip the first n molecules in SD file
--records <n> check only the first n records for field names
--noempty do not import empty molecules
--setchiralflag set chiral flag for MDL file formats.
--diff do not import ANY molecules, only output
--duplicates write duplicate molecules to output
--nonduplicates write non-duplicate molecules to output
--output redirect output to a file (otherwise stdout)
Options for export:
--where <condition> where clause. Example: --where "cd_id<1000"
Options for miscellaneous table management operations (command 'm'):
--add-ctcolcfg <opt> add Chemical Terms expressions to existing user-
defined columns. <opt> has the same format as the
argument to the --ctcolcfg parameter of the 'c'
(create table) command. The columns specified
must have no Chemical Terms expression
assigned to them.
--set-ctcolcfg <opt> set Chemical Terms expressions to existing user-
defined columns. <opt> has the same format as the
argument to the --ctcolcfg parameter of the 'c'
(create table) command. The columns specified may
have Chemical Terms expressions already assigned
to them.
--del-ctcolcfg <opt> remove the assignment of Chemical Terms expressions
from existing user-defined columns. <opt> is
a semi-colon-separated list of column names.
--set-abs-stereo (true|false)
set the Absolute Stereo table option.
--set-dupl-filtering (true|false)
set the Duplicate Filtering table option.
--set-use-tautomers (true|false)
set the 'Duplicate search uses tautomers' table option.
Note: Recalculation is needed after the change.
--set-switchoff-prots (true|false)
set the 'Switch off all protections for tautomer
duplicate checks' table option.
Note: Recalculation is needed after the change.
Examples:
$ jcman c strdata --coldefs ", name CHAR(200), stock INTEGER"\
--fplength 256 -l joe --driver com.mysql.jdbc.Driver\
--dburl jdbc:mysql://localhost/mydb -s
$ jcman c ctcols --coldefs ", logp numeric(18,9), rotbl_bnd_cnt numeric(1,0)"\
--ctcolcfg "logp=logp();rotbl_bnd_cnt=rotatableBondCount()>4"
...
$ jcman a str3d str3d.sdf.gz
$ jcman a str3d str3d.sdf.gz --connect "MOLNAME=NAME;PH=PH_VAL"
$ jcman t --rowcount
Precalc. Precalc. Precalc. Precalc. Precalc.
Table name Version Rows recommended version status valid invalid rows
1 str3d 5020100 198556 N
2 testdata 5020100 4260 Y 5020100 READY Y 16
$ jcman t ncidata
Column name Type name
1 cd_id LONG
2 cd_structure BLOB
...
18 cd_fp16 LONG
$ jcman m strtable --add-ctcolcfg "logp=logp();rotbl_bnd_cnt=rotatableBondCount()>4"4. Connecting to Databases
After starting JChemManager the "Connecting to a Database" dialog box appears. The dialog box can also be displayed by selecting the Connect icon on the tool bar of JChemManager. Tooltips, linked documents under ? icons, and warning messages in case of wrong or incomplete input can help the data entry in this dialog.
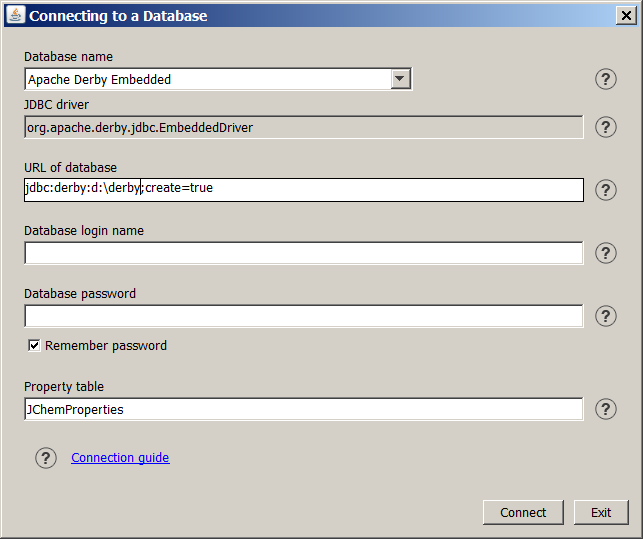
JChem uses the JDBC protocol to connect to relational databases. Before trying to connect, make sure that the appropriate JDBC driver is included in the CLASSPATH environmental variable. To establish a JDBC connection the following parameters have to be set:
Database name:
-
The Database name list provides database systems supported by ChemAxon. Select the adequate database system from the list or select Other in case of using other database than the listed ones. The following databases are supported by ChemAxon:
JDBC driver:
A Java class name, the entry point of the name of the JDBC driver. The appropriate driver of the selected database appears in this field. In case of Other database the driver must be specified in this textbox. If you would like to use an ODBC driver, enter sun.jdbc.odbc.JdbcOdbcDriver. See FAQ for more details on JDBC and ODBC drivers.
URL of database:
A JDBC URL provides a way of identifying a database so that the appropriate driver will recognize it and establishes a connection with it. At the first connection the URL format of the selected database system appears here. The parts highlighted by yellow color must be modified according to the local conditions. Some examples can be seen here .
In the case of an ODBC driver, the full syntax is
jdbc:odbc:<data-source-name>[<attributes>]where each attribute has the following form
;<attribute-name>=<attribute-value>For other common URL formats please click here.
Property table:
Enter the name of the property table. See JChem database concepts for the principles of JChem property table.
The default value of Property table is JChemProperties. Since version 1.6 default value can be changed by the user to support flexible multiuser capabilities and to support the same user to work within the same databse with more than one environment defined by different Property tables.
Login name:
Enter a user ID needed to connect to the database. If a login name is not needed, then leave the field empty.
Password:
Enter the password for the login name. If you want the system to save the password in the .jchem file for later use, check Remember password .
After filling the form and selecting Connect , the system attempts to connect to a database using the settings entered.
The above settings are stored in the .jchem file located under "chemaxon" or ".chemaxon" in the home directory of the user who connected to the database. At the first connection, a new table called JChemProperties (or the name you specify) is generated in the database, which contains parameters of structure tables.
5. Creating Structure Tables
Structure tables contain chemical structures and associated data, including both data used by the JChem system internally and custom, user defined data (static/imported or calculated). For more information about JChem table structure, see JChem database concepts.
To create a structure table select the Create icon. The Create a Table dialog box appears.
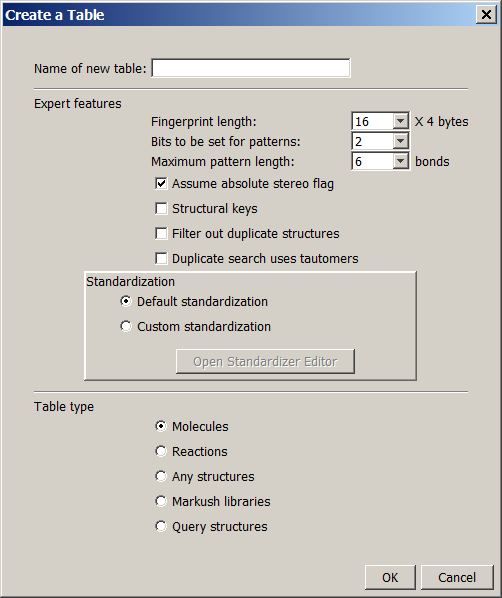
Parameters to be specified:
Name of new table:
The name of the table to be created in the database that was specified at the Connecting to a Database dialog box.
Fingerprint length*:
Specify the number of INTEGER columns that will contain the chemical hashed fingerprints of the molecules. Higher number provides better screening performance for substructure searching, but too many columns may significantly increase the size of the structure cache.
Bits to be set for patterns*:
The number of bits to be set for each substructure pattern in the chemical hashed fingerprints. 1 or 2 bits for a pattern are the best choice.
Maximum pattern length*:
The maximum length of linear substructure patterns used for chemical hashed fingerprints.
Assume absolute stereo flag:
If checked, all query and target structures are treated as absolute stereo. This setting can be changed later without recalculating the table.
Structural keys:
You can specify here a fix set of structures in a file that will be used as structural keys. The fingerprint will be extended with the appropriate number of integer columns to provide 1 bit for each structure.
Filter out duplicate structures:
If checked, JChem will filter out the structures which are already imported to the database with the same topology.
Warning: with a slight possibility concurrent import sessions may insert duplicated structures. See note about commit interval at Setting options.
Duplicate search uses tautomers:
If checked, tautomers are considered during duplicate search. Enabling this feature increases import time. This option is described in detail in the JChem Database Concepts.
Custom standardization:
You may specify a custom standardization XML for the structure table. You have to recalculate the table if you want to change the configuration. You can either select a standardization XML by clicking the 'Browse' button, or you can create your own with Standardizer Editor. Loading or saving a configuration in Standardizer Editor will set the target XML file being used as the custom standardization for the newly created table.
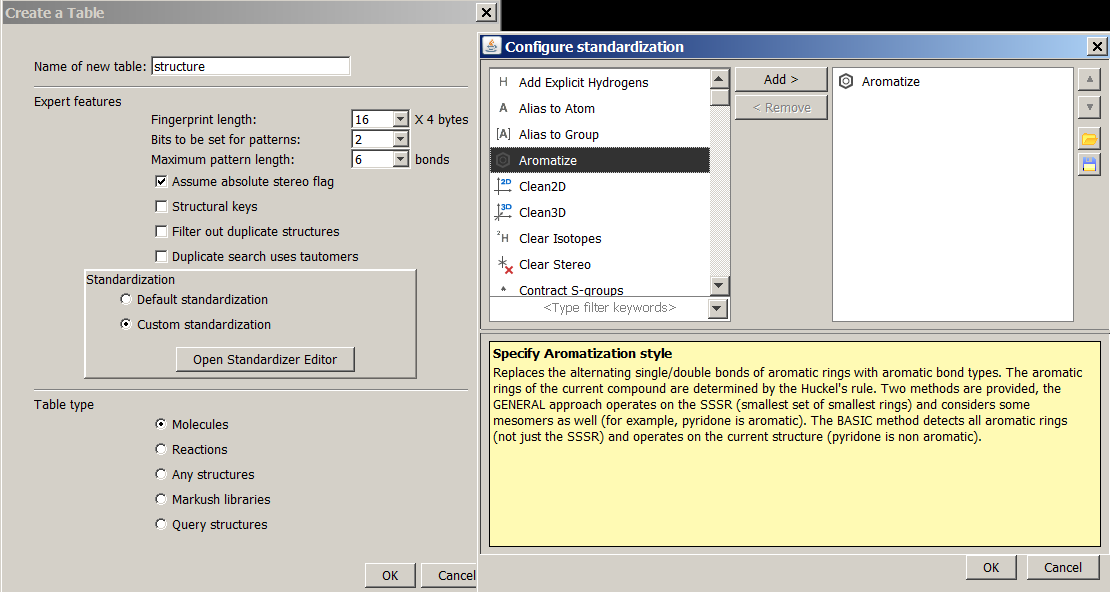
Table type:
-
You can select from the following table types according to the desired scope of use:
-
Molecules (default): For the storage of specific structures, like single molecules, mixtures, salts and polymers.
-
Reactions : Table for storing single step reactions.
-
Any structures : All types of structures are allowed, but no structure type-specific searching takes place (e.g. similarity values for reactions will not distinguish reactants, products and reaction centers).
-
Markush libraries : Table for storing markush structures. (this table type is not allowed for Ms Access dbms)
-
Query structures : Table for storing query structures. Typically used for superstructure search. Note: SMILES string imported into this table will be interpreted as SMARTS.
See more information at JChem Database concepts.
Compatibility notes : Tables created before JChem version 3.2 will be treated as "Any structures" to maintain previous behaviour. The default type for new tables is "Molecules".
*See the Chemical hashed fingerprint documentation about the optimization of these parameters. Statistics about fingerprint darkness of existing tables can also be obtained via running jcman s <table_name>. If you haven't made any previous testing, use the defaults that are optimized for typical compounds of pharmaceutical interest. The default values differ according to table type.
After pressing Ok in the Create a Table dialog box, the SQL statement for creating the structure table is displayed in the Create Table Statement dialog box.
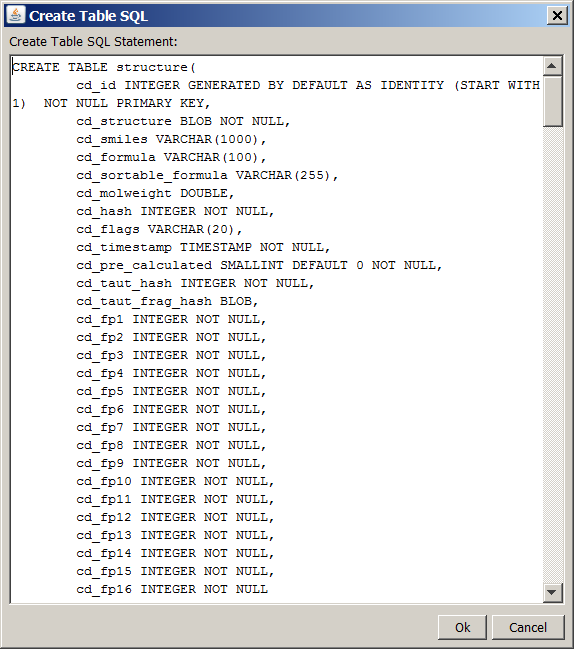
If you would like to add more columns, modify the SQL statement (though this can also be done later in most RDBMS-s).
If you would like to have Chemical Terms calculated columns (additional columns, the values of which are automatically calculated based on Chemical Terms expressions), you can specify them at the bottom of the dialog along with their respective Chemical Terms expressions. (The columns appearing in the bottom of the dialog as Chemical-Terms-based columns, must be also specified in the CREATE TABLE statement above. If you add columns later using your RDMBS and decide to make them Chemical-Terms-based columns, you can configure the Chemical Terms expressions for the new columns using the command line version of JChem Manager.)
NOTE:
-
The definitions of additional columns should start after the definitions of fix columns.
-
The name of extra columns should not start with "cd_".
There is a default limit on the length of the field cd_smiles for most RDBMS-s. If the majority of your molecules' SMILES representation is longer than this limit (in case of HUGE molecules), the search process can become slower. In this case you may try to increase the limit.
After selecting Ok, the SQL statement is executed.
For each structure table, the fingerprint properties are stored in the JChemProperties table. If the RDBMS supports schemata, username is also attached to the table name in the name column of the property table, like in the following example:
|
prop_name |
prop_value |
|
table.CDUSER.STRUCTURES.fingerprint.numberOfBits |
512 |
|
table.CDUSER.STRUCTURES.fingerprint.numberOfEdges |
6 |
|
table.CDUSER.STRUCTURES.fingerprint.numberOfOne |
2 |
6. Importing Structure Files
Import formats in JChemManager:
-
MDL Molfiles (a file containing concatenated Molfiles is created)
-
MDL Rxnfiles (a file containing concatenated Rxnfiles is created)
-
Daylight SMILES files (each line in the file is a SMILES string)
When the Import icon is selected, the "Import" dialog box appears.
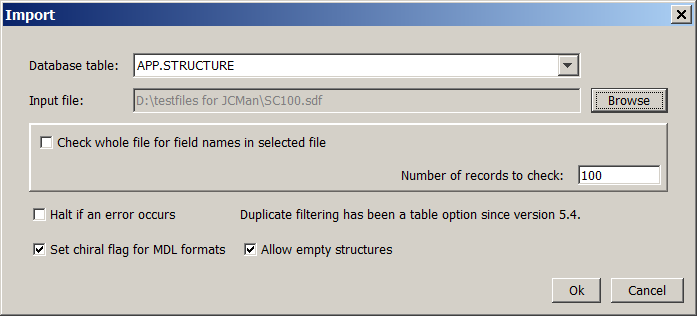
Specify the database table and the input file. The data fields in the file can be imported into the columns of the table.
To support connecting the corresponding field names and column names, the program will detect field names in the file. If the file is too big, checking may be time consuming. Use the Check whole file for field names in selected file check box and the Number of lines to check input box to decide whether you want the whole file or just a given number of lines to be searched for field names.
If an error occurs during import, the error message and the corresponding stack trace information is written to the standard error. Check the Halt if an error occurs box if you would like the system to stop if a molecule can not be imported.
If Allow empty structures is unchecked, JChemManager will not import empty structures (structures where the atom count is zero).
If Set chiral flag for MDF formats is checked, JChemManager will set the chiral flag (absolute stereo flag) for the imported structures.
In case of SMILES*, InChI, Mol2, Molfiles, RDfiles and Marvin Documents importing starts after selecting the Ok button.
For SDfiles, the "Connecting Fields" window appears, where you can connect the corresponding field names and column names.
*Some SMILES may contain additional data separated by whitespace from the structure string. The additional data columns are separated by tabulators ("\t"). In this case, the "Connecting Fields" dialog will also appear, the fields will be named as "field_0", "field_1" etc.
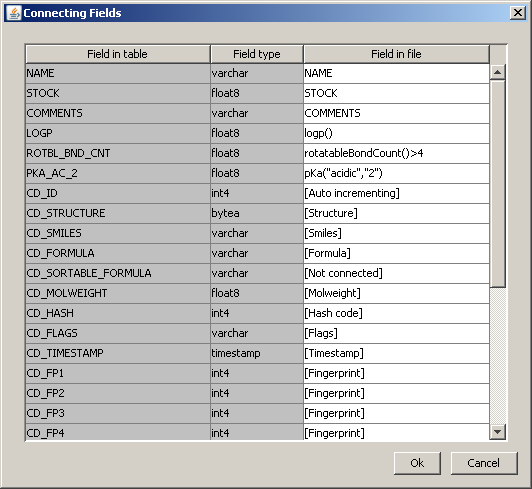
Clicking on a cell in the Field in file column of the window, a list box appears with the alternative field names. In the case of the cd_id column, Auto-incrementing can also be selected, which means that the value is increased by one after each new record. This works even if the RDBMS does not support auto-incrementing, because in this case JChemManager will take care of incrementing the value. (Some RDBMS-s that have the auto-incrementing feature for columns do not allow the explicit setting of cd_id.)
Importing starts after selecting the Ok button. A progress window displays the progress of the import.
7. Exporting Structure Files
Export formats in JChemManager:
-
MDL Molfiles (a file containing concatenated Molfiles is created)
-
MDL Rxnfiles (a file containing concatenated Rxnfiles is created)
-
Daylight SMILES files (each line in the file is a SMILES string)
When the Export icon is selected, the "Export" dialog box appears.
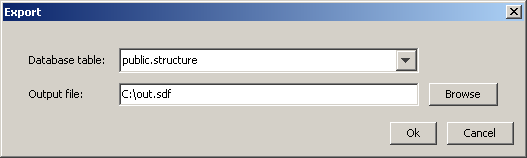 Specify the database table and the output file.
Specify the database table and the output file.
File format is determined from the extension:
|
Format |
Extension |
Examples |
|
MDL SDfile |
extension starts with "sdf" |
.sdf, .sdfile |
|
MDL Molfile |
extension starts with "mol" |
.mol, .molfile |
|
MDL RDfile |
extension starts with "rd" |
.rd, .rdf |
|
MDL Rxnfile |
extension starts with "rxn" |
.rxn, .rxnfile |
|
SMILES |
extension starts with "smi" |
.smi, .smiles |
|
InChI |
extension is "inchi" |
.inchi |
|
Mol2 file |
extension is "mol2" |
.mol2 |
|
Marvin Document |
extension is "mrv" |
.mrv |
After pressing the Ok button, the next dialog appears.
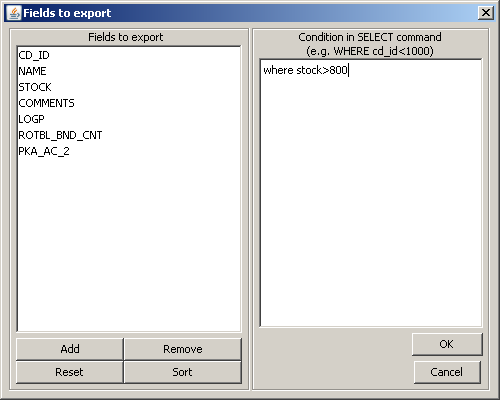
On the left panel, you can specify which fields to export in the case of formats that support additional data. By default, cd_id and the additional fields are selected (fields not beginning with "cd_"). You can add more fields by pressing the Add button. To remove one ore more fields, select them and press the Remove button. You can restore the default setting by pressing Reset. The Sort button arranges fields according to the original order in the database rows.
On the right panel you can specify a criteria for molecules to be exported in the form of an SQL WHERE statement .
Exporting starts after selecting the Ok button. A progress window displays the progress of the export.
8. Deleting Structure Tables
When the Delete icon is selected, the "Delete" dialog box appears.
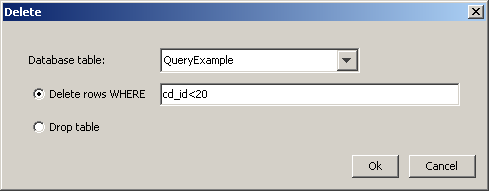
In this dialog you can
-
Delete rows from a structure table: You may specify which rows should be deleted in a where clause. If the field is left empty, all rows will be removed.
-
Drop a structure table: All settings referring to the deleted table are also removed from the JChemProperties table.
9. Modifying Structure Tables
Custom (non-cd) columns and Molecular Descriptors can be added to and deleted from a chosen structure table.
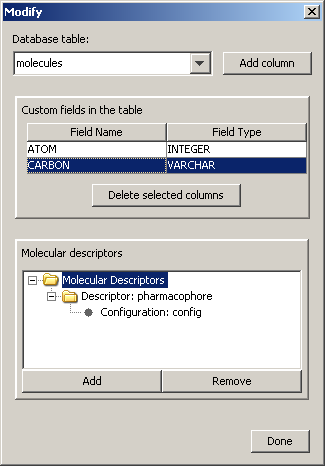
The structure table can be specified by the drop-down list found on the top of the pane. In the center there is a table which contains the custom field name and the type currently defined in the chosen structure table. To delete a custom field, select the row and click on the 'Delete selected columns' button. Molecular Descriptors and their configurations can also be seen on a separate pane. A custom column can be created by clicking on 'Add column' button.
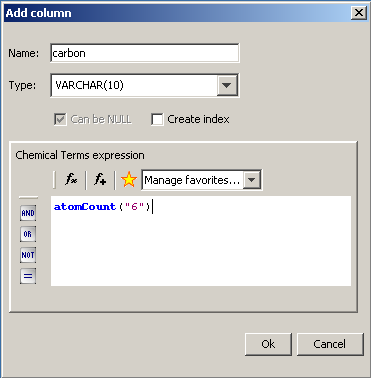
After pressing the "Add column" button, the next dialog appears. In this pane, the column name, column data type and its parameters can be defined with an optional chemical terms expression. Index can also be created for the column. When finished, click on the 'Ok' button to add the database field and after that you can add other fields by following the same process. Finally, click on the 'Done' button in the Modify dialog.
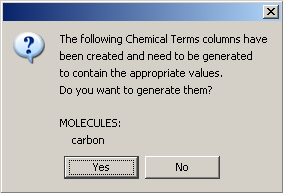
If any new columns containing chemical terms expression were added, a message dialog will appear to ask whether you want to generate the values of the newly created fields. If the 'No' button is selected, the columns will remain empty. If the 'Yes' button is selected, a progress bar will be displayed showing the status of the generation until the process is done. Chemical Terms values can be calculated or recalculated later as described here.
To add a Molecular descriptor, select the root element of the 'Molecular Descriptors' tree and click on the 'Add' button. Then the following dialog will pop up.
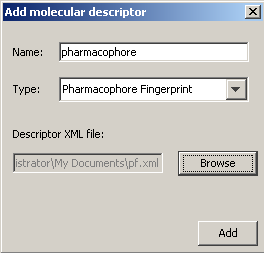
Here you must type the descriptor name, which can be anything. Then you should select the descriptor type and the descriptor XML file, which contains information about the custom fingerprint generation rules. You can define some additional configuration by clicking on the descriptor's name in the Molecular Descriptor tree. Give a name to the configuration and specify the XML file. By clicking on the 'Remove' button you can delete the currently selected descriptor or configuration in the Molecular Descriptor tree.
Updating and inserting rows in structure tables is not yet supported in JChemManager. However, custom applications built using the JChem Class Library can perform these operations. See the examples included with the package.
10. Changing Table Settings
The table options dialog can be reached from the File -> Table Options dialog of JChemManager.
Change the table name in the combo box on the top to view / edit settings for other tables.
Changing some settings (e.g. standardization, tautomer duplicate search) requires the recalculation of the table. This will be performed after pressing the "Ok" button. The recalculation can take considerable amount of time depending on the size of the structure tables and other factors.
NOTE: you can make changes for multiple tables, your changes will be stored when selecting other tables. The actual changes in the database and the recalculation (if needed) will take place for all tables after pressing the "OK" button.
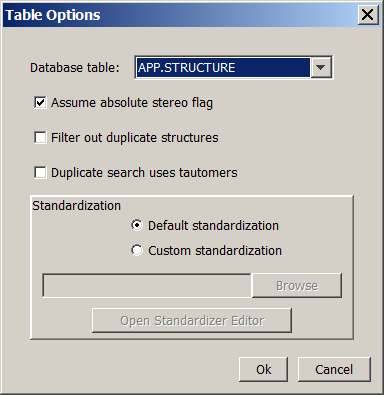
If "Assume absolute stereo flag" is set for a table, all query and target structures are treated as absolute stereo ("chiral flag" in MDL files).
Changing this setting does not require recalculation.
You can specify whether duplicate structures are filtered out during the import process or not.
Changing this setting does not require recalculation.
You can also specify if tautomers are considered during duplicate search. Enabling this feature increases import time.
Changing this setting requires recalculation.
You can also change the standardization of the tables.
Changing this setting requires recalculation.
11. Setting Options
When the Options icon is selected, the "Options" dialog box appears. The options set here are stored in the property table.
NOTE: If you are using multiple property tables these options should be set for each property table individually.
Advanced:
You can set advanced options in the second tab.
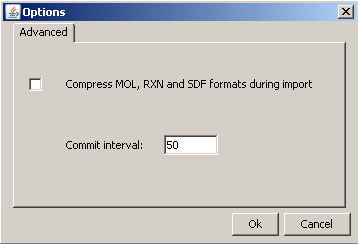
-
Compression: Certain file formats can be compressed before storing the structure source in the cd_structure field in the database. This has the following benefits:
-
Saves disk space in the database.
-
The compressed files can be directly sent to Marvin applets for display (text-based compression is used). This can reduce the download time for generated web pages.
-
Sometimes forced compression can be harmful, for example, when third-party software which cannot interpret compressed ChemAxon format need access to the structures. The compression is disabled by default, so all new structures are inserted without compression.
-
Commit interval: A database option, which defines the batch size for import. Your compounds will be committed in accordingly sized batches to the database during the import process. The value of setting may effect the speed of import and the efficiency of duplicate search in case of parallel import (running in different Java virtual machines). If this value is increased the speed of import will increase but there will be a slightly bigger chance of importing duplicates in spite of filtering (if a duplicate comes in process A which was not committed in process B yet).
12. Upgrading Database
After the installation of every major version of JChem and in certain cases after the installation of minor versions of JChem, you have to upgrade your database in order to make the database consistent with the new software version. Modification of database structure and of calculated data are executed during the upgrade process. There are two possible ways to upgrade database.
-
Start JChem Manager and connect to the appropriate database. 'Altering tables?' dialog will appear if the new JChem version contains changes in the database structure and lists the tables needing upgrade. Click on 'Proceed'. All listed tables will be recalculated.
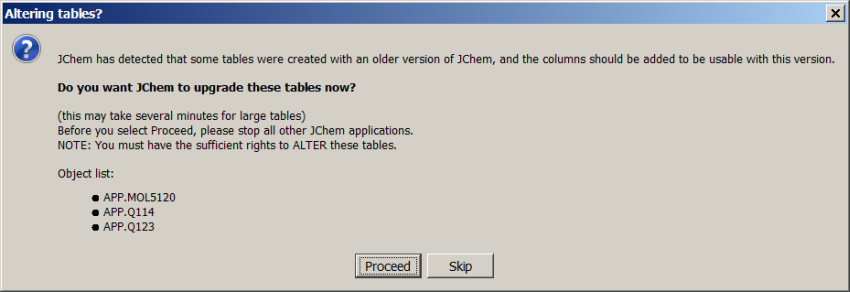
In the next step, 'Recalculating structure tables?' dialog will appear offering the recalculation of calculated values or Chemical Terms data in the database. Click on 'Proceed' if you want to run recalculation. Please be aware that recalculation can be run in a separate operation as described here.
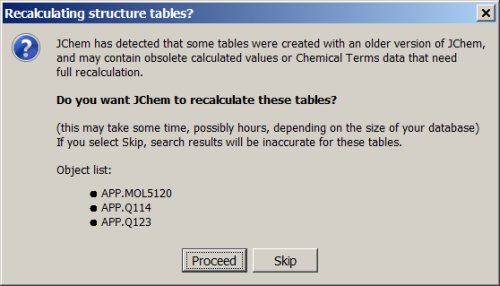
-
From command line:
jcman u upgrades database structure and recalculates old
tables if necessary 13. Recalculation of Fix Columns
When a new version of JChem is released usually the calculated data in the structure tables have to be refreshed to be consistent with the new version. Normally this is offered by JChem Manager when connecting, see above, but in some cases one may want to initiate recalculation by hand.
To recalculate fix columns, select the File -> Recalculate menu option, the "Recalculate" dialog will appear:
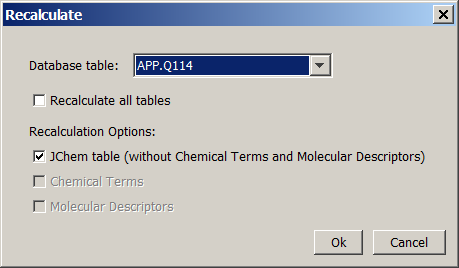
Three recalculation options exist. It is possible to recalculate either the standard JChem table fields, or the Chemical Terms fields of the JChem table, or the Molecular Descriptors which are stored in different tables, or even all of them. The recalculation options can be chosen with checkboxes, which are disabled if a given option is not possible for the selected table(s), e.g if there are no Chemical Terms defined. You can select one table, or recalculate all tables.
You can also recalculate the tables from command-line.
14. Recalculation policy
In which case is recalculation necessary
When upgrading to a new JChem version, either in JChem Manager or in command line mode, the user will be asked whether to recalculate some of the existing structure tables or not. In general the following rules are applied to decide whether to require recalculation or not when a new JChem release is coming.
-
Major versions (e.g. JChem 5.2.) always need full recalculation including Chemical Terms calculations.
-
Minor versions (e.g. JChem 5.2.1) need recalculation when there are modifications in the implementation that cause serious alterations in molecules' attributes or parameters (e.g., fingerprints).
-
There is a way to recalculate only the columns that contain Chemical Terms calculations. If it is necessary for a new release, but full table recalculation would be superfluous.
-
There is a way to recalculate everything except Chemical Terms columns. If it is necessary for a new release, but full table recalculation would be superfluous.
Recalculate Chemical Terms columns manually
Chemical Terms columns can be recalculated manually using the API, or the jcman in command line mode. In the API recalculateCTColumns method can be used, which is part of the UpdateHandler. The following line should be executed in command line to recalculate all Chemical Terms columns for a structure table.
jcman r --ct <tableName>
Table version details
For table version number one rule is applied: they must follow each other in an ascendant order which makes the recalculation check simpler. The version of one particular table can be viewed using the
jcman t [--rowcount]command for all structure tables, or
jcman t <tableName>for one particular table. Number 5020105 could be an example for table version. The first five numbers here define the JChem version 5.2.1 while the rest of this sequence is reserved for the possibility of further identification. The current JChem and table version can be retrieved executing the
jcman vfrom 5.3. In older versions, type
java -jar jchem.jarcommand in the lib directory, where jchem.jar can be found.
14. Precalculation
Concept
Recalculation process can be long sometimes, and since it is running on the live system it is not recommended to modify the molecules or the table structure until the recalculation has not been finished. Precalculation is implemented to make this pending time as short as possible. Precalculation process runs in the background, and precalculated data are stored in temporary tables. When finished, you can copy these data to the structure tables with one step. Meanwhile some molecules or the table structure may still change of course. In first case modified molecules should be recalculated after precalculated data have been applied (copied) but it will be automatic and will take much less time than recalculating the entire table. Structure change of the precalculated JChem table will invalidate the former precalculation so it has to be restarted again.
Note: When updating from a JChem version (older than 5.3) which doesn't implement precalculation, you should only use precalculation, if you are able to ensure that the precalculated table won't change between the precalculation process and the data copying.
Getting information about table precalculations
You can precalculate those tables which need recalculation according to the current version of JChem. You can get information about the need of precalculation, the status and the valdity of a previously started precalculation process by using
jcman t [--rowcount]If precalculation is recommended for a table but there is not precalculated data available with the corresponding version of JChem, or the data are not valid or there are too many changed (invalid) rows since the last precalculation, we suggest you to run the precalculation process on that table.
How to precalculate
You will need precalculation when you are upgrading your JChem from a previous version to a newer one. If you want to run precalculation you will need to install the new version because you have to use the precalculation process of this. During and after the execution of this process the older version can be used so the work does not need to be stopped on JChem databases. To run precalculation you should type
jcman kcommand for all structure tables, or
jcman k <tableName>for one particular table.
Note: Precalculation is only possible on tables which need recalculation according to the current version of JChem. Precalculation is not possible on tables which need upgrade (e.g., because of changes in database structure).
It is also possible that for some reason you would like to remove a temporary table created by the precalculation process. This operation can be perfored using the
jcman k --remove <tableName>command where tableName is the structure table which the temporary table belongs to.
Applying precalculated data
When you are starting JChem from GUI or using the command line jcman u option, JChem will automatically detect if there are complete and valid precalculated data available. In this case it will ask whether to apply these to structure tables or not. This will happen before JChem requires table recalculation and you can skip this step by answering No.
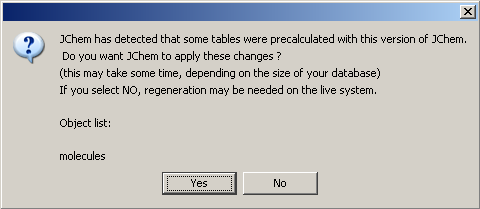
Otherwise precalculated data will be copied from temporary table to JChem table. It is possible that some molecules have been changed since the last precalculation. In this case a recalculation process is started for only these structures after precalculated data have been successfully copied. However, usually recalculation time of these rows must be much shorter than recalculation of the whole table. Of course, structure tables which already have the appropriate data gained by the way of precalculation won't appear later in the list of tables that need to be recalculated according to JChem.
Database support
Precalculation is supported on the following DBMSs now:
MySQL 4.0.4+
Oracle 9+
MSSQL Server 2005+
PostgreSQL 8+