ChemAxon's range of database products include JChem Base, JChem Cartridge for Oracle and Instant JChem. JChem Base provides the main chemical database intelligence and search engine, and is the basis of the other two products. The cartridge offers an Oracle SQL interface for JChem Base and other ChemAxon products, and Instant JChem is an all-in-one desktop chemical database application. This chapter describes the main concepts of JChem Base, which therefore are also relevant for the understanding of JChem Cartridge andInstant JChem.
Contents
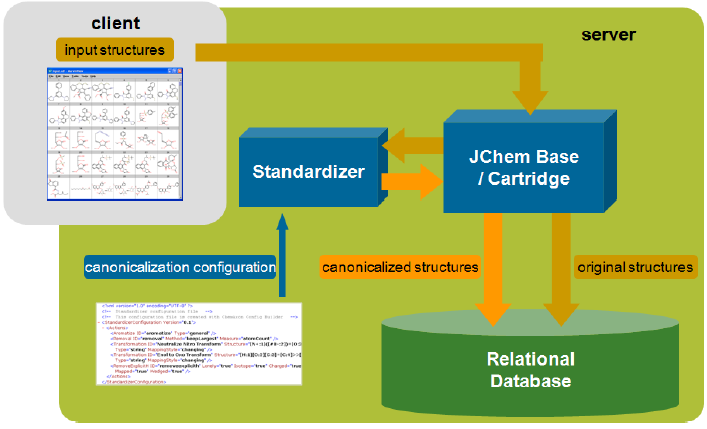
JChem Base architecture
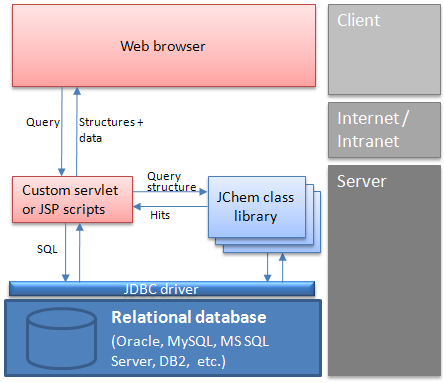
Web architecture: A typical interaction between a client and the database
-
Using a web browser, the user enters a structure into MarvinSketch applet.
-
A custom script (or servlet) for substructure/similarity searching is activated, which
-
Connects to a database through JDBC.
-
Searches in a table containing structures.
-
Creates a list containing the ID numbers of found structures.
-
-
The script retrieves mixed structural and non-structural data by SQL SELECT statements, using the hit ID numbers and tables or views in the database.
-
The script creates the page that displays the retrieved data in the client's browser using MarvinView applet .
The user manipulates the data, etc.
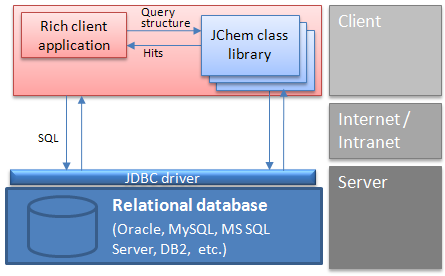
Rich client architecture: A typical interaction between a client and the database
Another solution is a two-tier architecture, where the client Java or .NET application uses JChem Base and JDBC API to interact with the database. In this case, chemical structure input and output may use Marvin Sketch and View beans components embedded into the client application.
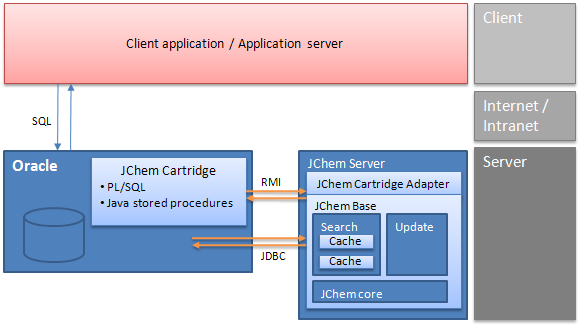
JChem Cartridge architecture
In case of the cartridge, the client application or application server communicates through SQL only, and all internal JChem Base operations are hidden. For efficiency reasons, the JChem Cartridge itself uses a JChem computation server that may reside on a dedicated server. More details can be found in the JChem Cartridge Developers Guide.
Instant JChem architecture is described in the Instant JChem documentation.
Table types
There are different structure table types available in JChem, depending on the desired structure content. The table type determines the checks at table import and influences certain searching operations on the table.
-
Molecules (default): This table type stores specific structures, like single molecules, mixtures, salts, coordination compounds and polymers. Supported search types: substructure, full structure, full fragment, duplicate, superstructure, and similarity search. For example, the following structures may be stored in molecule tables:
|
|
|
|
|
|
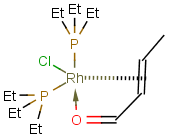
-
Reactions : Table for storing single step reactions. Only specific reactions which have neither query features nor query properties can be imported. Supported search types: substructure, full structure, full fragment, duplicate, superstructure, and similarity search. For similarity searching, it can use reactant, product or reaction similarity metrics (see details here). For example, the following reaction structure may be stored in a reaction table:
|
|
-
Markush libraries : Table for storing Markush structures. (This table type is not allowed for Ms Access DBMS.) Supported search types: substructure, full structure, full fragment, and duplicate search. See more information about the capabilities of these tables in the JChem Query Guide.
|
|
|
|
-
Query structures : Table for storing query structures. Typically used for superstructure search. Note: SMILES strings imported into this table will be interpreted as SMARTS. Standardization of the inserted structures is described in the standardization documentation of the query guide. Supported search types: superstructure search and duplicate search. For more information about available query features, see the JChem Query Guide. Query tables guarantee that all query features of stored structures are correctly handled during superstructure search.
|
|
|
|
|
|
-
Any structures : All types of structures are allowed. Supported search types: substructure, full structure, full fragment, duplicate, superstructure, and similarity search. However, no structure type-specific searching takes place in every cases (e. g. similarity values for reactions will not distinguish reactants, products and reaction centers; only the scaffold fragment of an R-group structure target is taken into account in substructure search). For example, the following structures may be stored in "Any structure" type tables:
|
|
|
|
|
|
|
|
|
Compatibility notes : Tables created before JChem version 3.2 will be treated as "Any structures" to maintain previous behavior. The default type for new tables is "Molecules".
Table type can be specified at table or index creation. (See, for example: JChem Manager or index creation in JChem Cartridge.)
JChem table structure
Structure tables contain chemical structures and associated data, including both those used by the JChem system internally and custom, user defined data. (User defined data may be any information related to the chemical structure: name, external id, physico-chemical properties, etc. Any number and type of user defined data can be added to JChem tables (within the limits of the underlying RDBMS) and can be standard (static) or calculated columns). The following columns are used by JChem internally. They are added at table creation. User defined columns can be added at table creation or any other time later.
cd_id (JDBC type: INTEGER)
-
Provides a unique identifier of the compound. If no value is specified for cd_id during the insertion of new structures, then the value is incremented automatically. A database index is automatically created for this column at table creation.
cd_structure (JDBC type: LONGVARBINARY)
-
Stores the structure in the original input format. It is used for displaying the structure and, in some cases, for searching (only when cd_smiles is not available). MDL Molfiles and SDfiles can be stored in compressed Molfile (csmol) form; the uncompressed form is stored by default. See Setting options in the Administration Guide.
cd_smiles (JDBC type: VARCHAR(1000)) or
cd_smarts (JDBC type: LONGVARBINARY) or
cd_markush (JDBC type: LONGVARBINARY)
-
These columns store the standardized structure in a compact format, allowing efficient caching and hence fast structure searching. (If this representation of the structure is larger than the maximum length of the column or cannot be represented for any other reason, then NULL is stored and the cd_structure field is used during the search.)
-
cd_smiles is used for Molecule, Any and Reaction table types, and contains ChemAxon Extended SMILES formatted structures.
-
cd_smarts is used for Query table type, and contains ChemAxon Extended SMARTS formatted structures.
-
cd_markush is used for Markush table type, and contains compressed Marvin documents of the internal Markush representation.
cd_formula (JDBC type: VARCHAR(100))
-
The molecular formula of the molecule, eg C7H6O2. The atomic symbols are in Hill Order: C is listed first, followed by H, followed by the remaining elements in alphabetical order. If the molecular formula is often used for searching, it is advised to create a database index on this column.
cd_sortable_formula (JDBC type: VARCHAR(255))
-
A transformed cd_formula (see above), which is available for correct alphanumerical sorting of formulas. (For example, C4H10 should precede C12H26 since 4 is smaller than 12, but the simple alphanumerical ordering of strings would result the opposite order.) In the sortable formula column, all numbers in the formula are left padded with leading zeros up to a constant length of 5.
cd_molweight (JDBC type: DOUBLE or FLOAT)
-
The molecular weight. If the molecular weight is often used for searching, it is advised to create a database index on this column.
cd_timestamp (JDBC type: TIMESTAMP)
-
The date and time of the insertion or the last update of the chemical structure in the row.
cd_hash (JDBC type: INTEGER)
-
A hash code of the chemical structure. It is used for duplicate search and in case of full structure search when no query features are specified on the query. It allows a rapid pre-filtering before atom-by-atom search. A database index is automatically created for this column at table creation.
cd_taut_hash (JDBC type: INTEGER)
-
A hash code of the generic tautomer of the chemical structure. It is used for tautomer duplicate search and in case of tautomer full structure search when no query features are specified on the query. It allows a rapid pre-filtering before atom-by-atom search. A database index is automatically created for this column at table creation.
cd_taut_frag_hash (JDBC type: VARCHAR(1000))
-
A list of hash codes for the different fragments of the chemical structures' generic tautomers. It is used during tautomer full fragment search when the query contains no query features for a rapid pre-filtering (screening) before atom-by-atom search.
cd_screen_descriptor (JDBC type: VARCHAR(1000))
-
This column contains descriptor data that are used in the screening phase of the search.
cd_flags (JDBC type: VARCHAR(20))
-
This column contains some special information stored in flags.
cd_pre_calculated (JDBC type: SMALLINT)
-
This column stores whether the table is in precalculated state or not.
cd_fp1, cd_fp2, cd_fp3, ...cd_fpn (JDBC type: INTEGER)
-
The fingerprints of the chemical structures stored in several INTEGER columns. It contains chemical hashed fingerprints and optionally structural keys. (If the table is configured that way.) Fingerprints are used during substructure and similarity searching in the fast screening phase. For reaction tables the reaction fingerprint of the reaction structure is stored instead to allow different reaction similarity search types.
JChem table names
JChem tables can only be referenced by their names OR by maximum one synonym in place of the table name. The use of more than one synonym is not supported. This limitation does not concern JChem Cartridge.
Update Log (UL) tables
For each new structure table an accompanying "myTableName_UL" table is also created. These tables are used for refreshing structure cache in concurrent environments. If an insert, update, delete operation is performed, it will be logged in the _UL table. The next search can update the structure cache incrementally based on these logs.
JChem property table
The JChem property table contains information about JChem's tables, registration information and further details about the database. Simply, this table identifies a JChem "environment" or "configuration". The default name of the table is "JChemProperties".
The JChem property table contains key-value pairs, like a property file or configuration file. The JChem Manager and Instant JChem applications and JChem Cartridge create and alter JChem Property Tables automatically. The JChem property table should only be edited by JChem applications or through the JChem API or JChem Cartridge operators/functions.
The property table contains these columns:
|
Column Name |
Description |
|
prop_name |
Keys that are used to access the value. |
|
prop_value |
The value of the property. |
|
prop_value_ext |
Used if the value property is too large for prop_value. |
Only one property value column (either prop_value or prop_value_ext) should be in use at any time. The other column should be null.
Relevant methods for creating a property table, checking for its existence, and adding, setting, or deleting properties can be found in the DatabaseProperties API.
There can be one or more property table for a database, located under the same or different schemas, if the database supports it. This can be used to create a multiuser database environment.
Search types
One major purpose of JChem tables is chemical structure search that can be combined with data search and is highly customizable. The following search types are available in JChem databases. Please click the links in the titles for more information.
-
This search type can be used to decide equality of molecules. It is used during duplicate filter import. All structural features (atom types, isotopes, stereochemistry, query features, etc.) must be the same for matching two chemical structures, but for example coordinates and dimensionality are usually ignored.
Nomenclature: In JChem versions prior to 5.2, this search type was called "perfect search". In other cheminformatics toolkits or cartridges this functionality may be called exact structure search.
-
Chemists are most often interested in this search type that decides whether a molecular structure contains a specific subgraph. Sometimes not only the chemical subgraph is provided, but certain query features also that further restrict the structure to search. If special molecular features are present on the query (eg. stereochemistry, charge, etc.), only those targets match which also contain the feature. However, if a feature is missing from the query, it is not required to be missing (by default). For more information, see the JChem Query Guide.
-
A full structure search finds molecules that are equal (in size) to the query structure. (No additional fragments (e.g. salt) or heavy atoms are allowed.) Molecular features (by default) are evaluated the same way as described above for substructure search.
Nomenclature: This search type was called exact search in JChem versions prior to 5.2, but was renamed to reduce confusion. (Note that this search type is NOT the same as the exact search of several other cheminformatics tools or cartridges, where it is used for finding duplicates. This latter functionality is called duplicate search in our terminology.)
-
Full fragment search is a combination of substructure and full structure search: the query must fully match to a fragment of the target. Other fragments may be present in the target, but they are ignored. This search type is useful to perform an "full structure search" ignoring salts or solvents stored with the main structure in the target.
Nomenclature: This search type was called exact fragment search in JChem versions prior to 5.2.
-
Superstructure search is the opposite of substructure search; it searches for those target molecules which can be found in the given superstructure query. For superstructure search, typically, the target structures with query properties are stored in Query structures type database tables. In "Query structures" type tables the default search type is superstructure search; substructure search is not allowed in them.
-
This search type is used to retrieve structurally similar chemical structures. By default, it uses the Tanimoto metric of chemical hashed fingerprints, but other screening configurations are also available by the JChem Screen integration. In this latter case, additional descriptor tables need to be added to the database, that link to the JChem table.
The JChem Query Guide describes each search type in more detail.
Search options
In addition to the above search types, there are many search options that modify structure search behavior. The most important options are listed below, the full and detailed list can be found in JChem Search Option Guide. Please click the links in the titles for more information.
-
This search option can instruct the search engine to look for all tautomer forms the query, as generated by the Marvin plugin Tautomers.
-
These search options allow a choice between several levels of strictness in matching bond types, especially regarding aromaticity. The higher the level is, the more tolerant the bond matching becomes.
The table below summarizes the vague bond levels.
|
Vague bond level |
Description |
|
Level 0 (off) |
Does not perform vague bond matching. |
|
Level half (default from version 15.9.14) |
Handling of 5-membered rings with ambiguous aromaticity |
|
Level 1 (default in versions prior to 15.9.14) |
Handling of 5-membered rings with ambiguous aromaticity, 1-atom-long aromatic ring ligands and bridging bonds between two aromatic rings become "or aromatic" |
|
Level 2 |
All query ring bonds become ″or aromatic″ |
|
Level 3 |
All query bonds (ring and chain) become ″or aromatic″ |
|
Level 4 |
Ignore all bond types |
-
This search option specifies how stereochemistry should be evaluated:
-
On (default): When the query does not contain stereo information, the hits will include results both with and without stereo information. Otherwise, the stereo information is taken into account during the search.
-
Exact: All stereo information is tested for equality, meaning that a non-stereo query only matches non-stereo targets
-
Diastereomer: retrieves stereo isomers where tetrahedral stereo information is present on the same stereo centers, but their configuration (parity) is arbitrary.
-
Off: All stereo information is ignored
Charge, isotopes, radical, valence settings
These search options specify how different atomic properties should be evaluated. Each of them has three settings. In the following the charge option is described, but all others of these options work the same way:
-
By default , an uncharged atom matches both charged and uncharged atoms and a charged atom only matches charged ones.
-
In exact charge mode, an uncharged atom only matches the uncharged atoms and a charged atom only charged ones.
-
In ignore charge mode, the charge is not checked during searching.
-
Searches can include extra conditions formulated in the Chemical Terms language. Chemical Terms is a chemistry language which allows users to formulate complex chemical questions, expressions and rules. Chemical Terms can contain references to functional groups, other structural elements and physico-chemical properties. The filter expressions are evaluated on the fly, but the Chemical Terms calculated columns are used if the column definition is part of the filter expression.
Combining structure search with other (non-structure) conditions
-
Non-structural conditions can be added to the database search by specifying an SQL statement through the filterQuery property. In case of the Cartridge, another solution is to combine JChem operations with other conditions in the WHERE clause of the SQL SELECT statement.
-
JChem Search Options Guide summarizes all available search options.
The structure cache
To boost the speed of searching JChem caches fingerprints and structures in the application's memory space. (In case of a web application, the application is usually an application server. In case of the Cartridge, it is the JChem server. In rich client applications, including Instant JChem, the structure cache is created on the client machine.)
The structure cache is stored in a static pool, therefore a structure table is only cached once within the same Java Virtual Machine (JVM). When structure tables change between search operations, the structure cache is incrementally updated to ensure minimum overhead. Introduced in JChem 5.3.2, cache registration helps the load and update process.
The build-up of the cache can take considerable amount of time and normally occurs once. You can do it directly, or it is done automatically, when the first search is started.
Depending on the number of molecules in the database, the size of the fingerprints, and the average molecule size, structure caching can have significant memory needs. Typically one million drug-like structures consume around 100 MB memory in the structure cache. JChem Base Performance Information contains more information about this subject.
To speed up descriptor-based similarity searching, it is recommended to cache the descriptor data. Caching of descriptor data is introduced inJChem 15.4.6. Caching can be enabled/disabled by setting sysprop.descriptor.caching.enabled system property to true/ false (in JChem Oracle Cartridge add this property to <jchem_home>/cartridge/conf/jcart.properties file). The current default is false, which may be a subject of change. The used descriptor data of a given table is loaded upon the first search, which means that it is significantly slower than the subsequent ones. The unused descriptor data are not loaded even if their table is searched. Descriptor data caching results in about an order of magnitude speed-up (depending on database access speed) while the storage requirement is roughly the table's cache size for every descriptor. Descriptor cache is unloaded by any update operation and the cache unload command (Java: CacheManager.unloadCache, JChem Oracle Cartridge: jchem_core_pkg.unload_cache).
Fingerprints, optimization of fingerprint parameters
JChem base uses different kinds of fingerprints for speeding up structural searches (via an initial fingerprint screening phase) and performing similarity searches. Fingerprints are bit strings that encode structural features present in the molecule. Different fingerprint types are used:
-
Chemical hashed fingerprints are used for most table types. These fingerprints are created by enumerating all linear patterns and rings (up to a predefined size) in the chemical structure, and the fingerprint bits are set using a hashing function.
-
Reaction fingerprints are used for reaction tables. These contain different chemical hashed fingerprint sections, to allow different reaction similarity methods.
-
Structural keys are optional additional bits appended the fingerprints relating to static patterns. A fix set of structures can be specified in a file that will be used as structural keys. The chemical hashed fingerprints will be extended with the appropriate number of integer columns to provide 1 bit for each structure. Important considerations related to structural keys:
-
If a substructure search is run against the structure table and the query structure is identical to one of the structural keys, the time of the search will be close to zero. This is because the substructure search was already performed at import, and JChem only has to check whether the specified bit is set to 1. This is useful if you frequently run substructure searches on the table using the same set of query structures.
-
If the query is not part of the structural key set, these keys are also considered for substructure and superstructure searches. Do not expect a major improvement in the effectiveness of screening in this case though, since the chemical hashed fingerprints are already very effective for most query structures.
-
During similarity search the structural key part of the fingerprint is not considered (dissimilarity is only calculated from the chemical hashed fingerprint part).
-
The speed of the import will slow down depending on the number of specified keys.
-
The required memory for the structure cache will increase with the increased number of fingerprint columns.
-
It must be taken into account that there are some query features which may cause loss of hits when used as features in structural keys. Wrong features are:
-
charge (when ignoring charges in the search)
-
isotope (when ignoring isotopes in the search)
-
aliphatic (A - does not have aromatic bond)
-
not member of a ring (R0)
-
-
The section about Chemical hashed fingerprints describes also how fingerprints can be optimized for good search performance.
Standardization
To ensure that structure search results are correct, the query and the database molecules must share a similar representation. This is achieved automatically through table standardization in JChem databases. For examples refer to Standardizer examples.
The database molecules are standardized during structure import into a JChem table (and also during structure update). First the original source of the chemical structure is stored in the cd_structure field, which can then be used for displaying and export purposes. The standardized form is then stored in the cd_smiles field in a compact format. This representation is used by the search process. All additional structure-dependent data (fingerprints, molecular weight and formula, Chemical Terms calculated columns) are also calculated from the standardized form. In case of JChem index in the Cartridge, this process is done during index creation (and during structure insert/update in an indexed structure column), and the standardized form is stored within the index.
Query structures are standardized automatically before the search.
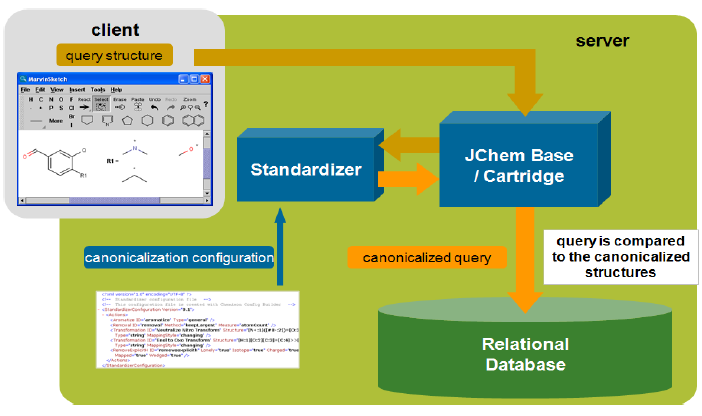
There are two types of standardization in the database:
-
Default standardization: By default, the bonds of aromatic systems are replaced with aromatic bonds and explicit hydrogen atoms are transformed to implicit ones when possible. This standardization is adequate in most simple cases.
-
Custom standardization: In some cases custom standardization is necessary, e.g. if nitro groups in the input structures are represented in two different forms. One can define custom standardization rules with a Standardizer configuration (XML or action string). The custom configuration can be specified at table or index creation. Custom standardization requires a Standardizer license.
This demo animation shows standardization setup inInstant JChem, and the following figure illustrates the Standardizer configuration builder and an example transformation that can be achieved using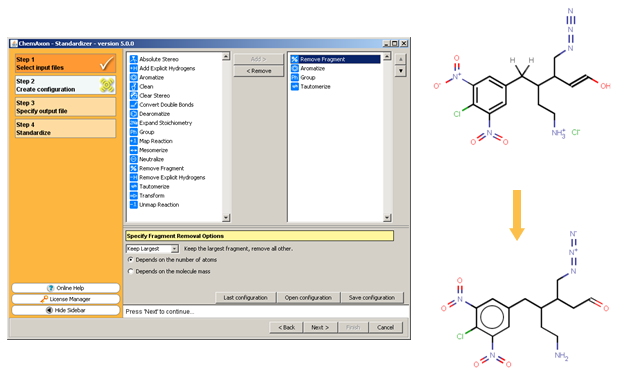
-
Standardizer:
For more information, see Standardizer configuration, Standardizer examples in Standardizer's User Guide.
Chemical Terms calculated columns
JChem database products uniquely allow the storage of a wide range of automatically calculated chemical properties in JChem tables and JChem indices. These properties are stored in Chemical Terms calculated columns that can be added at table creation or any other time later.
Calculated columns are automatically computed when a structure is inserted into the structure table or updated. The data to be calculated is defined by a Chemical Terms expression for each calculated columns. This language contains many structure-related functions, including the whole range of ChemAxon property calculations. A few examples for possible Chemical Terms column definitions are given below.
-
logP()
-
mass()
-
name()
-
molString('smiles')
-
rotatableBondCount()
-
(mass() <= 500) && (logP() <= 5) && (donorCount() <= 5) && (acceptorCount() <= 10)
Calculated columns can be created using Instant JChem, JChem Manager and JChem Cartridge: for JChem tables and JChem index. The figure below illustrates the calculated column creation dialog in Instant JChem, which contains a set of often used template expressions.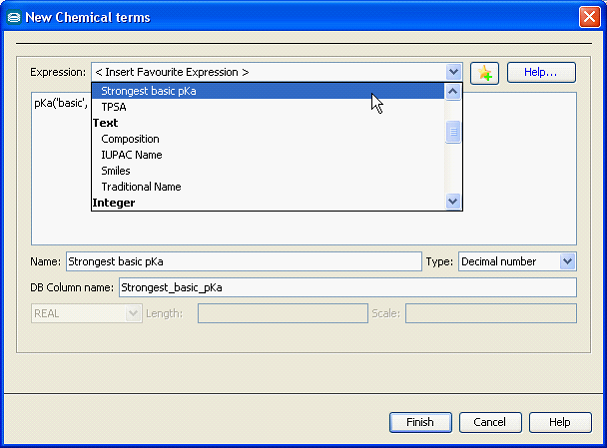
Handling of tautomers
Tautomers are structural isomers of organic compounds that are in dynamic equilibrium due to the migration of a proton. There are various solutions for handling tautomers in JChem.
For duplicate and full searches, tautomer search is executed using generic tautomers for 'any' and 'molecule' type tables. This way no enumeration of possible tautomers is needed; on the other hand, the hash code of the generic tautomer is calculated and stored in database which causes some overhead during database import.
1. Tautomer duplicate search table or index option
Tautomer duplicate search (tds) table option or index option. The tds option has the effect that - by default - duplicate search and duplicate filtering during import are executed considering tautomers as well.
Implementation and architecture notes for tables/index tables
-
cd_smiles contains the standardized version of the molecule, used by substructure, similarity, and full structure search.
-
cd_hash is calculated from the standardized molecule.
-
cd_taut_hash is calculated from the generic tautomer, however, the generic tautomer itself is not stored.
-
cd_taut_frag_hash is also calculated from the generic tautomer and contains a list of hash codes calculated for each fragment (per fragment hash code).
Generic tautomer search workflow in case of option 'tautomerSearch:On'
-
The query is standardized, and then its generic tautomer is created. In case of structures that can't have generic tautomer (e.g., polymers), the original molecule is taken. Tautomer hash code is calculated. In case of full fragment search the hash codes are calculated separately for each fragment (per fragment hash code).
-
Screening with tautomer hash code.
-
In case of duplicate and full structure searches the hash codes of the query and target generic tautomers are tested for equivalence.
-
In case of full fragment search the query per fragment hash code should be present among the target per fragment hash codes.
-
-
On the remaining records: read cd_structure, standardize, and then create generic tautomer.
-
The two generic tautomers (query and target) are checked with the appropriate (full/duplicate) atom-by-atom search. Extra settings are also used here, e.g., data S-groups of the generic tautomer are checked.
2. Tautomer search option
Tautomer option of structure searching
JChem uses the above generic tautomer approach in full, full fragment and duplicate searches.
In all other cases, it simply enumerates all theoretically possible tautomers of the query structure and searches them (as the query structure) one by one.
The interpretation of the default value of tautomerSearch (On or Off) depends on search type and on table option 'Duplicate search uses tautomers (tds)':
|
Search type |
Duplicate search uses tautomers |
|
|
Yes |
No |
|
|
duplicate |
On |
Off |
|
all other |
Off |
Off |
Stereo notes
See relating information about effects of tautomer search options on tetrahedral stereo matching and on E/Z stereo bond matching.
3. Canonical tautomer standardization
Canonical tautomer generation can be included directly into the standardization configuration of the table or index (tautomerize action). In this case, all search types will use the canonical tautomer. (Warning: the tautomerize action dearomatizes the structure, so an additional aromatize action must follow it.)
The canonical tautomer is generated by the ChemAxon Tautomerization calculation plugin based on empirical rules. It is a standard representation selected from all dominant tautomers of a molecule, so the same canonical tautomer relates to each of the dominant tautomers. The dominant tautomer model includes an energy (pKa) filter to remove the transformations that are unlikely in solution. Tautomerization also depends on the environment: phase (solid / solution), solvent, temperature, etc., but these are not considered in either of our methods.
The following poster has more information about the generation of all, dominant and canonical tautomers: Tautomer generation. pKa based dominance conditions for generating dominant tautomers.
4. Custom tautomer transformations in standardization
It is also possible to add your own transformation rules for separate tautomerizable functional groups as part of the table/index standardization. Examples can be found on Standardizer actions page .
Discussion of the methods
These options can be used together, though not always reasonable. For example 1 and 2 can be used for tautomer substructure search on a tautomer duplicate table.
Converting oxo-enol tautomers

XML action: <Transformation ID="enol"
Structure="[H:4][O:3][C:1]=[C:2]>>[H:4][C:2]
[C:1]=[O:3]"/>
action string: "[H:4][O:3][C:1]=[C:2]>>[H:4][C:2]
[C:1]=[O:3]"Converting enamine-imine tautomers
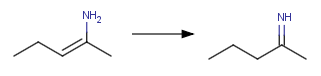
XML action: <Transformation ID="enamine"
Structure="[H:4][N:3][C:1]=[C:2]>>[H:4][C:2]
[C:1]=[N:3]"/>
action string: "[H:4][N:3][C:1]=[C:2]>>[H:4][C:2]
[C:1]=[N:3]"All four methods are suitable for duplicate search, but for substructure search there are different issues:
-
Option 1. is not suitable for substructure search because the generation of identical generic tautomers is assured only in case of full structures.
-
The canonical tautomer generation algorithm requires a full molecule to properly consider energetics and the local structural environment of tautomerizable functional groups. For this reason, option 3. is not ideal for substructure search.
Therefore, only solutions 2. and 4. are recommended for substructure searching.
Concerning search speed, solutions 1., 3. and 4. are the fastest to search, because all transformations are done at registration time. Solution 2. is much slower to search than all other options.
Registration (indexing) speed is fastest at solution 2. (No registration overhead.) Second fastest is solution 1. (Little registration overhead.) Solutions 3. and 4. are the slowest to register. (Depending on standardizer configuration complexity.)