Huckel Analysis Plugin
This manual gives you a walk-through on how to use the Hückel Analysis Plugin:
Introduction
The Hückel Analysis Plugin is able to calculate different structural descriptors for aromatic atoms.
Localization energies L(+) and L(-) for electrophilic and nucleophilic attack at an aromatic center are calculated by the Hückel method. A smaller L(+) or L(-) value means more reactive atomic location. Order of atoms in E(+) or in Nu(-) attack are adjusted according to their localization energies. The total π energy, the π electron density and the total electron density are also calculated by Hückel method. Depending on the chemical environment, the following atoms have optimal Coulomb and resonance integral parameters: B, C, N, O, S, F, Cl, Br, I.
All other atoms have a default, non-optimized parameter. Theoretical background is taken from Isaacs' book. Additional literature for the Hückel's parameters is Streitwieser's book.
The results appear in a new window, indicating the calculated values at the atoms in the aromatic ring. In the following example the first picture is the result of Aromatic E(+)/Nu(-) order calculation, while the second picture is the result of a Pi energy calculation.
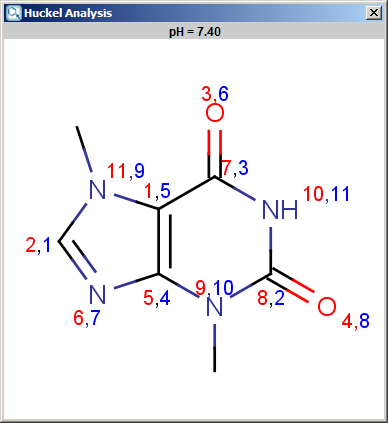
Fig. 1 Aromatic E(+)/Nu(-) calculation result
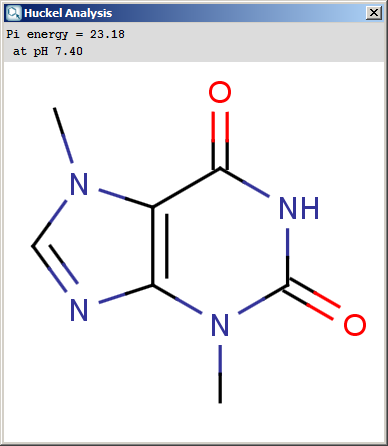
Fig. 2 Pi energy calculation result
Options
The following calculation parameters can be set in the Huckel Analysis Options window:
-
Decimal places: setting the number of decimal places with which the result value is given.
-
Type: sets the method of calculation. That could be:
-
HMO E(+)/Nu(-) order: numbers the aromatic atoms according to their probability of being attacked by electrophiles or nucleophiles.
-
HMO Localization energy L(+)/L(-): gives the localization energies of the aromatic center (dimension β).
-
HMO Pi energy: calculates the π energy of the aromatic ring(s) (dimension β).
-
HMO Electron density: calculates the π electron density.
-
HMO Charge density: calculates total charge density on the ring atoms.
-
Localization energy L(+)/L(-): the same as the HMO Localization energy L(+)/L(-), but based on experimental (reaction) data
-
Electron density: the same as the HMO Electron density, but based on experimental (reaction) data
-
Charge density: the same as the HMO Charge Density, but based on experimental (reaction) data
-
-
Subtype: E(+) or Nu(-): for E(+)/Nu(-) order and localization energy L(+)/L(-), the electrophilicity and nucleophilicity approaches can be selected (at least one of them). Results for E(+) are coloured red, and Nu(-) blue.
-
Take major microspecies at pH: calculates the values for the major microspecies at the given pH.
On the HMO method
The HMO is an abbreviation for the Hückel Molecular Orbital theory/method. The HMO calculation methods mentioned above (e.g. HMO Pi energy) uses α and β parameters based on literature data.
The non-HMO methods were developed for predicting reaction products in ChemAxon's Reactor. The α and β parameters used in these methods were determined to give the closest results to the expected experimental results in reaction product predictions with Reactor. Regarding the details of Reactor, see the reference article.
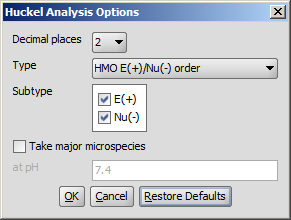
Fig. 3 Huckel Analysis Options window
References
-
Isaacs, N.S., Physical Organic Chemistry, John Wiley & Sons, Inc., New York, 1987, ISBN 0582218632.
-
Streitwieser, A., Molecular Orbital Theory for Organic Chemists, John Wiley, 1961, ISBN 0471833584.
-
Pirok, Gy. et al., Making "Real" Molecules in Virtual Space, J. Chem. Inf. Model. 2006, 46, 563-568