Contents
-
User defined operator (deprecated)
Introduction
JChem Cartridge adds chemical knowledge to your Oracle platform and you automatically take advantage of Oracle's security, scalability, and replication. You can search data by structure, substructure and similarity through extensions to Oracle's native SQL language. Chemical data can be easily inserted and modified using SQL procedures of the JChem Cartridge.
SQL operators of the Cartridge can be used to search for chemical data (molecules or reactions) more efficiently than it is possible using traditional mechanisms such as PL/SQL Stored Procedures. In order to avoid unecessarily slow searches, applying the jc_idxtype index type is required.
Architecture
High Level Overview
The chemical knowledge for JChem Cartridge comes from ChemAxon's core Java libraries. Oracle offers the possibility to use existing Java libraries by calling them from Java Stored Procedures. Java Stored Procedures run in a special, in many respects unconventional Java runtime environment which is tightly integrated with the Oracle RDBMS. One of the most important characteristics of the Oracle Java runtime environment is that Java libraries are run in a byte-code-interpreted mode. This aspect puts Java Stored Procedures at a serious disadvantage in terms of performance compared to modern Java runtime environments. These latter use sophisticated techniques to compile Java byte-code into native binary executable at runtime. Native executables can then be run much faster than Java byte-code.
The solution used for JChem Cartridge to compensate for the performance disadvantage with Oracle Java Stored Procedures is to perform the computation intensive functions in an external Java runtime environment capable of runtime byte-code compilation ("external" meaning: external to Oracle). We will call this external Java runtime environment (as well as the JChem Cartridge components running in it) JChem Server.
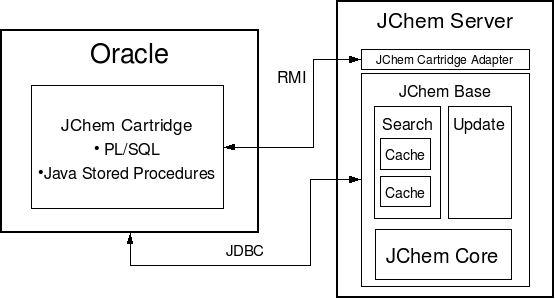
At an abstract level, the JChem Server comprises the following components:
-
JChem Base providing
-
the core chemical functions such as atom-by-atom graph search algorithms(JChem Core);
-
implementing advanced database search and update functions. (Search and Update engines which add an RDBMS-aware layer to the core JChem functions).
-
-
JChem Cartridge Adapter which translates JChem Base's external interfaces to the needs of JChem Cartridge.
For structured database access, the Search and Update engines use JDBC connection(s).
To reduce hard disk access and network round trip, the Search Engine extensively caches the chemical information stored in database tables. (How to configure memory for the search cache.)
The JChem Cartridge components (PL/SQL and Java Stored Procedures) which are located within Oracle, perform the following functions:
-
Interfacing with the Oracle Data Cartridge framework,
-
Performing simple tasks involving mainly pure database access,
-
Managing communication with the JChem Server.
The Oracle-resident Java classes of JChem Cartridge are packaged in jcart.jar.
Database Access Modes
The standard way for an Oracle Java Stored Procedure to access database objects is to use a JDBC-connection which has been pre-opened by Oracle in the context of the database session, in which invocation of a given stored procedure has been initiated.
However, as JChem Cartridge stored procedures make extensive use of the JChem Server for all kind of tasks to speed up computation, there are cases where it is much more efficient to access the database (as part of some sub-tasks) directly from within the JChem Server rather than relying entirely on stored procedures for database access. Relying entirely on stored procedures for database access would require moving (in some cases) a large amount of data back and forth between the JChem Server and the stored procedures which would have an obvious impact on performance (and in the case of earlier Oracle versions also on stability).
Direct database access from within the JChem Server thus involves opening a new database session (within JChem Server). This results in certain cartridge operations using two database sessions: the "original" (initiator) database session and an auxiliary database session. INSERT operations into regular Oracle tables as well as inserts into JChem tables using the jchem_table_pkg.jc_insert function are currently performed exclusively using the initiator session. A single session is also used for updates and deletes.
The dual-session access mode is used for searches that use the jc_idxtype operators in domain index scan mode (where the table is scanned using the jc_idxtype domain index). An exception is the duplicate search, when the requireCommit option is specified with the n value. Another (related) exception is the duplicate search performed to enforce uniqueness during structure insert either using jchem_table_pkg.jc_insert with the checkDuplicates option set to true or using regular INSERT into a regular structure table indexed with the duplicateFiltering index parameter. The functional equivalents of operators as well as the operators executed in "non-domain-index-scan" mode use one singe database session.
Dual-Session Database Access Example
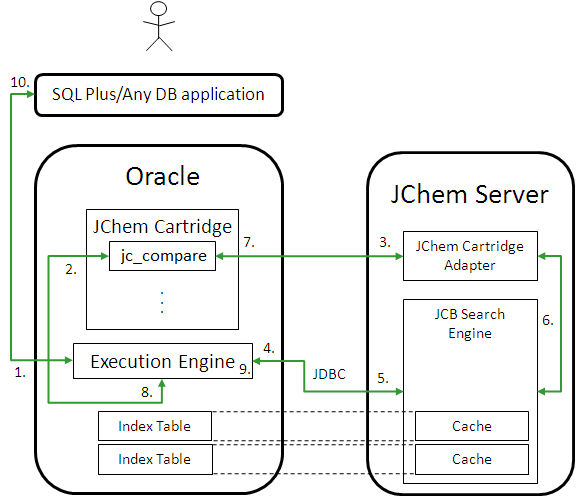
When you use a search operator (such as jc_compare) in the WHERE clause of a SQL query, the following execution path is traversed:
-
The application sends Oracle Server a SQL statement involving substructure search.
-
The Oracle Execution Engine processes the statement and the substructure search operator of JChem Cartridge is called with the appropriate parameters.
-
The parameters of the search are passed onto the JChem Server over RMI.
-
The Search Engine checks (over the JDBC connection) to see if the database table involved in the search has been modified since the corresponding structure cache was loaded the last time.
-
The structure cache is refreshed (over the JDBC connection), if necessary.
-
A pre-screening is used on fingerprints in the cache, then an atom-by-atom graph search on the structures from the cache.
-
The results are communicated over RMI back to the cartridge components located in Oracle.
-
The ROWIDs of the matching structures are returned to the Oracle Execution engine.
-
The Oracle Execution engine further processes the original SQL statement, if necessary.
-
Results are returned to the user.
Single-Session Database Access Example
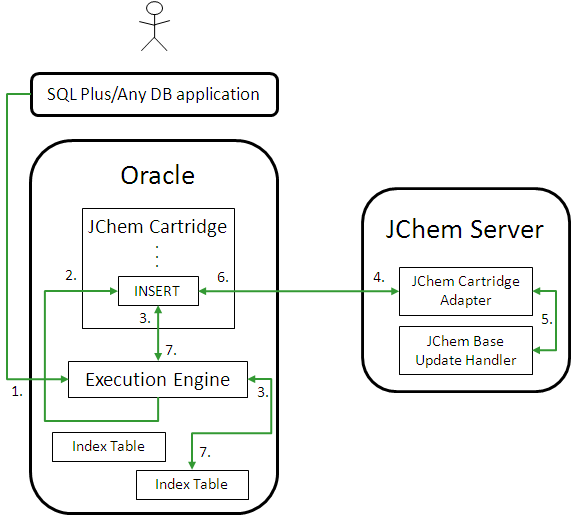
-
The application sends Oracle Server a SQL statement involving an INSERT (in the case of regular structure tables) or jc_insert (in the case of JChem structure tables).
-
The Oracle Execution Engine processes the statement and calls JChem Cartridge to either update the index table for the newly inserted structure (in the case of regular SQL INSERT) or directly execute the jc_insert function. (jc_insert includes both inserting the structure and updating the index information in the same JChem structure table.)
-
Additional information (Standardizer configuration, fingerprint generation rules) is fetched from the index table.
-
The molecular structure is passed onto the JChem Server over RMI along with the additional information.
-
JChem Base's Update Handler computes the chemical information to update the index table with.
-
The computed chemical information is communicated over RMI back to the JChem Cartridge components located in Oracle.
-
The chemical information is inserted into the index table.
Implications of the Dual-Session Database Access Mode
Using two separate JDBC connections for searching has the following implications:
-
User authentication information must also be maintained in the JChem Server.
-
Searching is performed in a separate database session and thus in a separate transaction context. The consequence is that modifications in the database are not reflected in search results until they are committed.
User Credentials
While it is currently impossible to retain "normal" transaction semantics with dual-session database access, beginning with JChem version 3.0.12 a simple mechanism is provided to allow to retain security semantics: i. e. to execute all database operations with the credentials of the initiating database session. The mechanism consists of
-
the JChem Server maintaining in transient form (in system memory) the user credentials (user name, and password);
-
propagate the user name of the initiating user for each call to JChem Server, so the proper credentials can be used to open the JDBC connection in the JChem Server on behalf of the user action "in Oracle".
A PL/SQL procedure jchem_core_pkg.use_password(password VARCHAR2) is made available for users to set their transient password in JChem Server. While the password is kept in JChem Server's main memory, it will be remembered and used until JChem Server is shut down. It is therefore recommended but not strictly necessary for users (or applications on their behalves) to call the use_password procedure at the beginning of each database session. (It must called once after each time the JChem Server is (re)started.)
If the password of a given user has not been set for JChem Server via the use_password PL/SQL procedure, the user credentials (if any) that were set as part of the JChem Server JDBC configuration will be used to open database connections in JChem Server.
jchem_core_pkg.use_password(password VARCHAR2) transmits the password to JChem Server via in plain text via a regular RMI call. If JChem Server and Oracle are running on different machines, malicious users may be able to sniff the passwords as it travels on the network. The necessary steps must be taken to prevent this (e.g. by limiting user's privileges on client machines so that they are unable to put their Ethernet card in promiscuous mode, or using a dedicated/closed network connection between the Oracle host and the JChem Server host).
Support For JChem tables
In addition to regular structure tables containing molecular structures, structure tables that have been created and populated by tools included in JChem Base can also be used with JChem Cartridge. The advantages of JChem tables over plain Oracle tables are:
-
inserts into JChem tables are faster than into plain tables,
-
searching in JChem tables are faster than in plain tables,
-
certain features (such as chiral flag setting for MDL files or duplication filtering) can be customized at a structure level for JChem table imports, while the same features can be customized at an index level granularity with plain Oracle tables. The disadvantage of JChem tables is that you often cannot use the standard SQL commands to manipulate/access them. The following limitations apply:
-
You have to use JChemManager to create and drop JChem tables.
-
You have to use either JChemManager or the jchem_table_pkg.jc_insert function provided with JChem Cartridge to insert into a JChem table.
-
You have to use either JChemManager or the jchem_table_pkg.jc_update procedure provided with JChem Cartridge to update structures in a JChem table.
-
You have to use either JChemManager or the jchem_table_pkg.jc_delete procedure provided with JChem Cartridge to remove structures from a JChem table.
From an architectural perspective, the main difference between using/indexing regular structure tables and using/indexing JChem tables is the following:
When you create a JChem Cartridge domain index on a regular table, the chemical information on the structures found in the table (called in this context base table) is stored in a separate table (called index table). Since the same chemical information which is stored in an index table is already present in a JChem table, the base table and index table are physically the same table in case of a JChem table. (Thus creating a jc_idxtype index on a JChem table is an instantaneous operation.) In the case of JChem tables, you have to create the JChem Cartridge domain index on the cd_structure column.
Extensible Index, jc_idxtype
The purpose of the jc_idxtype index type is to enable efficient search and retrieval functions. Currently the CREATE INDEX and the ALTER INDEX SQL statements are supported.
CREATE INDEX
The index can be created on a column of any table, provided that the column contains molecular structures in any of the supported representation. Use the following general statement to create the index:
CREATE INDEX <index name> ON <table> (<structure-column>) INDEXTYPE IS jc_idxtype PARAMETERS('param1=paramvalue1,param2=paramvalue2,...');The index name cannot currently be longer than 22 characters. The following optional parameters can be specified for both JChem and indices on regular structure tables:
-
JChemPropertiesTable is required for determining the properties of fingerprints during the queries. These properties, that are stored in the JChemProperties table, can be different for each JChem molecule table. If the JChemPropertiesTable parameter is not defined, the method uses the default property table name in the user's schema: JChemProperties.
Note that the user creating the index has to has to be granted INSERT, UPDATE and DELETE privileges on the JChemProperties table. In the case of the default JChemProperties table (being in the index creator's schema), these privileges are automatically available. In case the JChemProperties table is stored outside the index creator's schema, these privileges has to be explicitly granted.
The following option can be specified only for JChem tables:
-
RegenerateTable: specify this option if you want to create index on a structure table generated by an earlier, incompatible version of JChem. Trying to create an index on an incompatible JChem-table version without specifying this option will result in an error message. This option accepts no parameter value.
In addition to the preceding parameters the following can be used for indices on regular structure tables:
-
idxSubType [ structures | fp ]: specifies which one of the two kinds of currently supported contents are about to be indexed:
-
structures for chemical structures (default)
-
fp for chemical fingerprints Fingerprints (specified as idxSubType=fp) must be given as "bit strings", that is a series of "0" and "1" characters, representing individual bits in the fingerprint. All "bit strings" in the indexed column must be of identical size. The index parameter fp_size must be set to the length (measured in 32 byte long integers) of the "bit strings". During similarity search, queries must be specified using the same "bit string" format.
-
-
fp_size: the number of INTEGER columns (each INTEGER column is 32 bytes long) that will contain the chemical hashed fingerprints of the molecules. (For more details see the JChem Administration Guide.)
-
fp_bit: the number of bits to be set for each pattern in the structure.(For more details see the JChem Administration Guide.)
-
pat_length: the number of edges in a pattern.(For more details see the JChem Administration Guide.)
-
structureType: the type of the structures being indexed. The following structure types are currently valid:
-
anyStructures: All types of structures are allowed, but no structure type-specific searching.
-
molecules: Specific structures, like single molecules, mixtures, salts, polymers. Other structures (e.g. with query or Markush features or reactions) are not allowed in database (exception is thrown).
-
reactions: Index on single step reactions.
-
queries: Index on queries. Only superstructure, full and duplicate searches are allowed.
-
markush: Structures with markush features. Markush search license is required. For more information, see the JChem Developers Guide .
-
-
structuralfp_config: a select statement returning the structural keys.
-
std_config: the configuration for custom standardization. The standardizer configuration is used during structure search. See here how. When not specified, the standardization configuration set as a default property is used. When not specified and no default standardization property is configured, the built-in default standardizer configuration is used. For details see the Standardizer documentation. You can use either the XML format or the action string variant of the Standardizer configuration. Special care has to be taken when custom standardization (other than the build-in default) is used on the database table. The "aromatize" action should be present in the configuration, and it is safest to put it first. See also the examples below.
-
std_conf:sql: similar to std_config with the exception that instead of the Standardizer configuration itself, the value of this option must be an SQL statement returning the Standardizer configuration.
-
absoluteStereo [y|n]: if set to "y" all query and target structures are treated as absolute stereo. This setting can be changed later with ALTER INDEX. This setting can be overridden for individual searches with the corresponding option of the jc_compare operator.
-
haltOnError [y|n|nf|nfp]:
-
If set to "y" (the default value), indexing will immediately abort upon the first error.
-
If set to "n", indexing will not immediately abort with an error message on detection of a badly formed structure (or on a duplicate structure if the duplicateFiltering option is turned on). In case error occurred, indexing will abort after all structures were processed with a generic error message indicating the number of errors occurred. The errors ignored during indexing can be retrieved in JChem Server's logfiles.
-
If set to "nf" (never fail), the behaviour is similar to the "n" value, except that indexing will complete "successfully" in the sense that the index will be left in a usable state (as if no error had been occurred). This value must be used with particular caution, as only the log entries will indicate that not all structures have been included in the index data.
-
If set to "nfp" (never fail persist), errors related to individual target structures being indexed are ignored during the existence of the index – including errors during ALTER INDEX, INSERTs and UPDATEs. (Examples of the kinds of errors ignored are errors in the representation format of target structures or errors due to target structures being outside the input domain of specific features defined for the given index such as autocalculated Chemical Terms) The errors ignored are logged in either a default auto-managed error table or a custom error table which can be specified through the errorTableName index parameter. In contrast to the custom error table, the default error table is automatically managed along with the index (deleted, if the index is dropped, truncated if the table is truncated, etc). The name of the error table currently in use for a given JChem index can be queried using the following SQL:
select jchem_core_pkg.get_error_table_name(<index-owner>, <index-name>, <partition-name>) from dual(The <partition-name> parameter is typically null.) This parameter affects only indexing of the structure column and has no effect on DML operations.
-
-
duplicateFiltering [y|n]: if set to "y", indexing and INSERT will check whether the structure being indexed are a duplicate of a structure already indexed in the same column. If the new structure is a duplicate, an exception is thrown. (Specify the haltOnError parameter, if you want to avoid restarting indexing on each problematic structure in the column being indexed.)
-
TDF [y|n]: If set to "y", DUPLICATE search will consider tautomers as duplicates. If set to "y" and the duplicateFiltering option is also turned on, duplicate filtering will consider tautomers of the structure being checked on duplication as well. More details about the theory behind this option can be found in the JChem concepts chapter of the JChem Developers Guide.
-
exclusiveDF [y|n]: if set to "y" and the duplicateFiltering option is also turned on, only one database session will be given the chance at a time to insert or update the structure column in order to effectively serialize otherwise concurrent non-atomic duplicate search and update/insert operations. This means that once a database session starts inserting into (or updating) a structure column, other database sessions subsequently starting to insert into (or update) the same structure column will block until the first session commits. Concurrent searches are not affected: they can proceed as usual.
-
autoCalcCt=<column-type-definition>;<ct-expression> specifies Chemical Terms calculated columns : that the expression should be automatically evaluated on structures being indexed with the value being stored in a column of the specified type. The expression will be evaluated during CREATE INDEX for the structures already inserted and automatic calculation of the values during future structure inserts is arranged. The precalculated values will be used by jc_evaluate and jc_evaluate_x functions. (See example)
-
TABLESPACE to specify the table space where the database objects associated with the index should be stored. Note that this parameter applies only for objects specific to the index being created. Certain small-foot-print objects serving as schema-scoped meta data repository shared by multiple indexes (such as the JCHEMPROPERTIES table) are not affected. Use: TABLESPACE=<tablespace-name>.
-
STORAGE to specify any of the elements of the standard Oracle storage_clause. Use: STORAGE=<storage-clause-elements>.
-
threadCnt=<number-of-indexing-threads> to specify the number of threads used for indexing. This setting overrides the value of the indexingThreadsPerCall property in the jchem/cartridge/conf/jcart.properties file. If neither is set, as many indexing threads will be used as the number of cores available on the JChem Server host.
-
insSessCnt=1 to specify that only one database session (connection) is used to insert the generated index data into the database. If this parameter is not specified or is set to 0 (zero is the default value), each indexing thread will use a separate database session to insert the index data they generate.
-
If the chemaxon.jchem.cartridge.indexingIsAynch is set to true in the jchem/cartridge/conf/jcart.properties file, the current status of the index creation can be monitored via the v$session_longops table. The refresh period in miliseconds of this table can be set by the refreshPeriod parameter. The default value of the parameter is 1000 (1 second).
-
errorTableName to set the name of the error table . Logs the error messages only if haltOnError is set to "n" or "nf". If the errorTableName parameter is not set, the error messages are available only through the standard log. Here are some examples to create an index:
-
Create index with default parameters:
CREATE INDEX jc_idx ON jchemtest(structure_col) INDEXTYPE IS jc_idxtype; -
Create index on a column containing 1024 bit long fingerprints:
CREATE INDEX jcxextfp ON extfp(fp) INDEXTYPE IS pkovacs_priv.jc_idxtype PARAMETERS('idxSubType=fp,fp_size=32') -
Create index with default parameters:
CREATE INDEX jc_idx ON jchemtest(structure_col) INDEXTYPE IS jc_idxtype; -
Create index with default parameters on a JChem table for use with BLOB operators:
CREATE INDEX jc_idx ON jchemtest(cd_structure) INDEXTYPE IS jc_idxtype; -
Create an index with structural keys stored in the stkey column of the stkeys table:
CREATE INDEX jc_idx ON jchemtest(cd_structure) INDEXTYPE IS jc_idxtype PARAMETERS('sep=! structuralfp_config=SELECT stkey FROM stkeys'); -
Create index with Daylight-style aromatization:
CREATE INDEX jc_idx_2 ON jchemtest2(structure_col) INDEXTYPE IS jc_idxtype PARAMETERS('std_config=aromatize'); -
Specify that query and target structures must be treated as absolute stereo by default:
CREATE INDEX jc_idx_2 ON jchemtest2(structure_col) INDEXTYPE IS jc_idxtype PARAMETERS('absoluteStereo=y'); -
Specify that
-
the LogP of the structures being indexed be stored in a column of type numeric(30,15)
-
the rotatableBondCount be stored in a column of type numeric(10,0)
-
the pKa in a column of type numeric(30,15):
CREATE INDEX jcxautocalccttest ON autocalccttest(structure) INDEXTYPE IS jchem_cc.jc_idxtype PARAMETERS('sep=! autoCalcCt=numeric(30,15);logp()!autoCalcCt=numeric(10,0);rotatableBondCount()!autoCalcCt=numeric(30,15);pKa("acidic","2")')
-
-
Create index so that JChem stores metainformation about the index in a table called jchemprops:
CREATE INDEX jc_idx_2 ON jchemtest2(structure_col) INDEXTYPE IS jc_idxtype PARAMETERS('JChemPropertiesTable=JChemProps'); -
Create an index on a structure table generated by an earlier, incompatible version of JChem so that the table will be upgraded to the current JChem table version:
CREATE INDEX jc_idx_2 ON jchemtest2(structure_col) INDEXTYPE IS jc_idxtype PARAMETERS('RegenerateTable=true'); -
Create an index on the USERS tablespace with the storage_clause 'INITIAL 2M' using Daylight style aromatization:
CREATE INDEX jc_idx_2 ON jchemtest2(structure_col) INDEXTYPE IS jc_idxtype PARAMETERS('TABLESPACE=USERS,STORAGE=INITIAL 2M,std_config=aromatize'); -
The following example assumes that that the confaux table contains a valid Standardizer configuration in the line specified by the where clause:
CREATE INDEX jcxstdcnfsql ON pkovacsuser_trunk.stdcnfsql(structure) INDEXTYPE IS PKOVACS_TRUNK.jc_idxtype PARAMETERS('std_conf:sql=select value from confaux where confkey = ''std_config'',tableType=molecules')
ALTER INDEX
The following parameter can be specified for jc_idxtype indexes created on columns of regular structure tables:
-
addAutoCalcCt=<column-type-definition>;<ct-expression> adds a Chemical Terms expression which should be automatically evaluated on structures being indexed with the value being stored in a column of the specified type. The expression will be evaluated during ALTER INDEX for the structures already inserted and automatic calculation of the values during future structure inserts is arranged. The precalculated values will be used by jc_evaluate and jc_evaluate_x functions. (See example.)
-
delAutoCalcCt=<ct-expression> stops automatically evaluating the given Chemical Terms expression on structures being indexed and drops the column holding the already calculated values. (See example)
-
addDfltMdConf=<md-type-short-name>[:<md-name>] to add a molecular descriptor to an index with default settings where
-
<md-type-short-name> can be any MD type accepted by GenerateMD
-
<md-name> is optional; if not specified the name of the molecular descriptor will be <md-type-short-name>.
-
-
addMd=<configuration-locator> to add a molecular descriptor to an index; where <configuration-locator> can be either
-
a SELECT SQL statement returning rows of three columns with each row specifying a molecular descriptor configuration with
-
the first column containing the descriptor name,
-
the second column containing a descriptive comment and
-
the third column (of type either VARCHAR2, CLOB or BLOB) containing the actual configuration xml . Please, see the specifying parameters through a temporary database table for an example.
-
-
the path to "a metadescriptor file". Please, see the specifying parameters through files on a filesystem for an example.
-
-
delMd=<descriptor-name> to delete a molecular descriptor.
-
addMdConf=<descriptor-name>:<configuration-locator> to optionally add an alternative screening configuration to an already existing molecular descriptor. <configuration-locator> can take on the same forms as for the addMd parameter. See Add alternative screening configurations for more details.
-
delMdConf=<descriptor-name>:<screening-configuration-name> to delete an alternative screening configuration associated with a descriptor. Examples
-
Adds ECFP fingerprints to the index:
alter index jcxnci parameters('addDfltMdConf=ECFP')In order to perform similarity search on the ECFP fingerprints use the descriptorName search option:
select count(*) from nci where jc_dissimilarity(struct, 'C[C@H](CS)C(=O)N1CCC[C@H]1C(O)=O', 'descriptorName:ECFP') < 0.3 -
Adds the following Chemical Terms expressions to the list of precalcuated expressions:
-
the LogP of to be stored in a column of type numeric(30,15)
-
the rotatableBondCount to be stored in a column of type numeric(10,0)
-
the pKa to be stored in a column of type numeric(30,15):
alter index jcxautocalccttest parameters('sep=! addAutoCalcCt=numeric(30,15);logp()!addAutoCalcCt=numeric(10,0);rotatableBondCount()!addAutoCalcCt=numeric(30,15);pKa("acidic","2")')
-
-
Removes the following Chemical Terms expressions from the list of precalcuated expressions:
-
the LogP of to be stored in a column of type numeric(30,15)
-
the rotatableBondCount to be stored in a column of type numeric(10,0)
-
the pKa to be stored in a column of type numeric(30,15):
alter index jcxautocalccttest parameters('sep=! delAutoCalcCt=logp()!delAutoCalcCt=rotatableBondCount()!delAutoCalcCt=pKa("acidic","2")')
-
ALTER INDEX ... REBUILD
This command is supported only for indexes created on structure columns of regular structure tables. The equivalent functionality for JChem structure tables is available through JChemManager or Instant JChem.
When specified without parameters for a jc_idxtype index, this command will rebuild the index.
The following parameters are accepted for jc_idxtype indexes:
-
upgradeOnly If this parameter is specified with the value y, the index will be rebuilt only, if the index data has been calculated using a JChem version older than the current.
-
skiprecalc If this parameter is specified with the value y, index data will not be recalculated. Only the index's internal structure will be cleaned up base on parameters as were used (last time) to create (or alter) the index. Use this command with precaution, because it will clear the status flags of the index and an index with missing or fault data will be marked as valid.
-
skipAutoCalcCt If this parameter is specified with the value y, index data recalculation will not include the recalculation of the auto calculated values of Chemical Terms expressions associated with the given index. (Recalculation of the auto calculated values of any Chemical Terms expression associated with the given index can be achieved by first removing the Chemical Terms expression from the index, then readding it.)
-
absoluteStereo to specify/change default chirality.
-
TABLESPACE to specify the table space for the index. Use: TABLESPACE=<tablespace-name>.
-
STORAGE to specify any of the elements of the standard Oracle storage_clause. Use: STORAGE=<storage-clause-elements>.
-
skipRecalc to specify that the index data should not be recalculated. With this parameter, it will only be checked wether the structure and version of the specified index is consistent with the currently installed JChem Cartridge version. (This mode can also come handy, if you want to bail out from an alter index operation aborted due to a syntax problem with addAutoCalcCt.
-
std_config: specifies a new Standardizer configuration, if needed. See also std_config for CREATE INDEX.
-
std_conf:sql: similar to std_config with the exception that instead of the Standardizer configuration itself, the value of this option must be an SQL statement returning the Standardizer configuration.
-
haltOnError [y|n|nf]:
-
If set to "y" (the default value), index rebuild will immediately abort upon the first error.
-
If set to "n", index rebuild will not immediately abort with an error message on detection of a badly formed structure. In case an error occurred, index rebuild will abort after all structures were processed with a generic error message indicating the number of errors. The errors ignored during index rebuild can be retrieved in JChem Server's logfiles.
-
If set to "nf" (never fail), the behaviour is similar to the "n" value, except that index rebuild will complete "successfully" in the sense that the index will be left in a usable state (as if no error had been occurred). This value must be used with particular caution, as only the log entries will indicate that not all structures have been included in the index data.
-
-
refreshPeriod: similar to refreshPeriod parameter at CREATE INDEX.
-
errorTableName : similar to errorTableName parameter at CREATE INDEX.
Examples
-
Recalculate the index data:
alter index <index-name> rebuild; -
Move the index table for a jc_idxtype index to the SYSTEM tablespace, with storage_clause 'INITIAL 3M BUFFER_POOL KEEP':
alter index jcidx_nci_1k rebuild parameters('TABLESPACE=SYSTEM,STORAGE=INITIAL 3M BUFFER_POOL KEEP');
Index operators
Parameters representing target structures can be either a structure string in any format recognized by JChem or a column containing such structures.
Some of the parameters representing target structures can only be a structure string (literal-only target structures).
Parameters representing query structures can be in any format supported by JChem (like SMARTS, Molfile, Rxnfile, etc.). Starting with JChem 3.1.1, multiple query structures can also be specified for a single search operator at a time. The individual query structures are to be separated as specified by the given format (e.g. in the case of SMILES/SMARTS, SDFiles with a newline character). The search is performed for each individual query structure and the union of the hits is returned. (See also our Future Plans.) Multiple query structures are accepted only for operators evaluated in domain index scans. See the example for jc_compare.
Some of the operators and functions have an option list parameter. Option list parameters are of type VARCHAR2 and accept a list of options (specific to the given operator or function). The default separator between the individual options is the space character. In case the space character is not appropriate as a separator (because, for example, the value of any of the options itself contains space(s)), a custom separator string can be used by specifying the special option "sep=" as the first option followed by a string that will be used as the custom separator. The custom separator string must be delimited by a space at the end (the space will not be part of the separator string). If a custom separator string is specified (always at the beginning of the options string), each option following option must be delimited using the custom separator string.
Without creating a jc_idxtype index of a chemical table, operators occurring in expressions are evaluated for each row of the result. The operators will therefore throw an exception to remind you of a missing index.
If the value of the smiles column of the current row is NULL then a NULL value will be returned by the operator.
Examples:
SELECT cd_id, jc_compare(smiles, smiles,'t:s') FROM scott.jchemtable WHERE cd_id < 10;
SELECT cd_id, jc_tanimoto(smiles, 'CN1C=NC2=C1C(=O)N(C)C(=O)N2C') FROM scott.jchemtable WHERE cd_id < 10;
SELECT cd_id FROM scott.jchemtable WHERE jc_compare(smiles, 'CN1C=NC2=C1C(=O)N(C)C(=O)N2C','t:s') = 1;
SELECT cd_id FROM scott.jchemtable WHERE jc_evaluate(smiles, 'logd( "7.4" )' ) > 2;NOTE: Operators appearing in the WHERE clause are evaluated much faster with the use of the JChem index jc_idxtype .
Example:
SELECT cd_id FROM scott.jchemtable WHERE *jc_compare(smiles, 'CN1C=NC2=C1C(=O)N(C)C(=O)N2C', 't:s') = 1;
Functional equivalents
Each operator supported by the jc_idxtype index type have a functional equivalent in the jcf package. The name of the functional equivalents have the 'jc_' prefix of their operator peers removed. E.g. the functional equivalent of jc_molconvert is jcf.molconvert.
One possible use of functional equivalents is in SQL constructs where operators cannot be used such as embedded in another operator:
select jc_compare('c1ccccc1', jcf.molconvert('C1C=CC=CC=1', 'smiles:au'), 't:s') from dual;
Default properties
The behavior of operators and functions can be configured by using default properties and index parameters. Currently, two global properties are supported:
-
the Standardizer configuration called standardizerConfig, whose value can be a list of actions.
-
the absolute stereo flag called absoluteStereo, whose value can be false and true.
For example to use chemaxon-style aromatization as default:
call jc_set_default_property('standardizerConfig','aromatize:b');
The effect of setting this property will be two-fold:
-
Its value will be used to define the behavior in contexts where no index information is available (e.g. select ... from dual).
-
Its value will be used as the default when creating an index of jc_idxtype . You can override the default value for a given index by specifying the corresponding index parameter with a different value (the std_config index parameter for the standardizerConfig default property).
Default properties provide default values only to indexes that are created in the current user's schema. The scope of default properties is the schema. Each user requiring daylight style aromatization with dehydroganization must call
call jc_set_default_property('standardizerConfig','dehydrogenize:optional..aromatize');to initialize their environment. If the user does not specify their own default, a built-in default standardization will be used (which includes dehydrogenization and a Generic aromatization).
The property value can be up to 32766 charater long.
The valid operator comparison using the index are:
-
jc_compare(...) = 0
-
jc_compare(...) = 1
Deprecated operators:
-
jc_contains(...) = 0
-
jc_contains(...) = 1
-
jc_equals(...) = 0
-
jc_equals(...) = 1
jc_matchcount, jc_tanimoto, jc_dissimilarity, jc_molweight, jc_evaluate operators return real values. These index operators support the full range of comparison functions: ">", ">=", "<", "<=", and "=". Note that the "!=" comparison does not use the index and will use the functional form.
The jc_compare operator with t:s and t:d option parameters perform substructure search and duplicate structure search respectively. The operator take three parameters, the first is the smiles column, the second is the query structure in any format supported by JChem (like SMARTS, Molfile, Rxnfile, etc.), the third is an (option list) of search parameters. Substructure search (t:s option) returns the structures from the table, which contain the query as a substructure, duplicate search (t:d option) returns the ones that are equal to the query structure. If, for some reason, you need a different search behavior, have a look at other search options ofjc_compare.
SELECT id FROM JCHEMTEST WHERE jc_compare(smiles, 'CN1C=NC2=C1C(=O)N(C)C(=O)N2C', 't:s') = 1;The jc_matchcount operator performs substructure search and returns the occurrence of the query structure in the target structure.
NOTE: In case of some search options (e.g. substructure search) via the jc_compare and jc_matchcount operators, if the query structure is given in SMILES format it will be converted to SMARTS. See more here.
Similarity search using Tanimoto formula can be performed by using the jc_tanimoto and jc_dissimilarity operators. The return values of these operators are in the range of 0.0 - 1.0, inclusive.
SELECT id FROM jchemtest WHERE jc_tanimoto(smiles, 'CN1C=NC2=C1C(=O)N(C)C(=O)N2C') < 0.6;
SELECT id FROM jchemtest WHERE jc_dissimilarity(smiles, 'CN1C=NC2=C1C(=O)N(C)C(=O)N2C') > 0.4;Molecular weights are calculated by the jc_molweight operator.
Select id from JCHEMTEST where jc_molweight(smiles) < 23;Calculations can be performed by using the jc_evaluate, jc_compare operators.
SELECT id FROM jchemtest WHEREjc_evaluate(smiles, 'logp()') < 11;
SELECT id FROM jchemtest WHEREjc_evaluate(smiles, 'logd( "7.4" )' ) > 4.2;
SELECT id FROM jchemtest WHEREjc_evaluate(smiles, 'psa()') > 3;If two or more operators are used in the where clause of one select statement it is more efficient to use thejc_compare operator with a fiterQuery .
Note, that since the caching search engine uses its own JDBC connection to Oracle, the updates to the structure tables must be committed before searches include the recent changes in their results.
Index statistics
The jchem_core_pkg.get_idx_stats function can be used to to collect statistics on the chemical information in jc_idxtype indexes. The jchem_core_pkg.get_idx_stats function accepts three parameters:
-
the index owner's schema;
-
the second the name of the index;
-
the third the name of the index partition (for partioned local jc_idxtype indexes). Example:
select jchem_core_pkg.get_idx_stats('jchemuser', 'jcxnci_10k', null) from dualA sample output follows:
Statistics for table: JCHEMUSER.JC_NCI_10K -------------------- Row count: 10000 NULL SMILES count: 0 Average SMILES length: 27.66 Average compressed SMILES length: 12.96 Markush structure count: 0 (0.0%) Min. CFP bits: 0.78% cd_id: 2110 Max. CFP bits: 68.35% cd_id: 3035 Avg. CFP bits: 21.48% Chemical Fingerpint distribution: -------------------------------- 0% - 10% : 15.38 % 10% - 20% : 34.04 % 20% - 30% : 29.55 % 30% - 40% : 14.69 % 40% - 50% : 4.91 % 50% - 60% : 1.21 % 60% - 70% : 0.22 % 70% - 80% : 0.0 % 80% - 90% : 0.0 % 90% - 100% : 0.0 %
User defined function (deprecated in version 15.3.2)
JChem Cartridge supports the ability to execute external program objects written in Java. To make an external Java program available from SQL, a PL/SQL function (and an according operator) and a Java class has to be defined.
The PL/SQL function has to call the send_user_func function of the jchem_core_pkg PL/SQL package to send the data to the external Java program. This function has three VARCHAR2 type parameters:
-
the name of the external Java program's main class
-
separator string to separate parameters in the following list
-
parameters list separated by the separator string
For example the my_func PL/SQL function and the my_op operator to call the MyClass external Java class:
CREATE FUNCTION my_func (query VARCHAR2, param VARCHAR2) RETURN NUMBER AS
BEGIN
RETURN jchem_core_pkg. send_user_func ('MyClass', '{DELIM}', query || {DELIM} || param);
END;
/
show errors;
CREATE OPERATOR my_op BINDING(VARCHAR2, VARCHAR2) RETURN NUMBER
USING my_func;The external Java class has to implement the chemaxon.jchem.cartridge.JChemCartModule interface. This interface contains the doFunc function that performs the operation on the data was sent by the previously defined PL/SQL function. The doFunc function accepts a String array parameter. This String array contains the items of the parameter list that was defined in the third parameter of the send_user_func function. The doFunc function of the external Java class returns an object and sends it back to the PL/SQL function. The newly created Java class has to be found on the classpath of JChem Server.
For example the MyClass Java class:
public class MyClass implements chemaxon.jchem.cartridge.JChemCartModule
{
public Object doFunc(String[] args) throws Exception
String query = args[0];
String param = args[1];
Object result = null;
...
result = ...
return result;
}
}Make sure that the xclasspath property in the jchem/cartridge/conf/jcart.properties file is set to the parent directory of the MyClass.class file. Now the new function is ready to be called from PL/SQL, for example:
SELECT my_op (smiles) FROM jchemtable WHERE id < 11;
SELECT count(*) FROM jchemtable WHERE my_op (smiles) >= 4;
In the example above the doFunc function of the MyClass external Java class gets the value of the smiles column of the current row as parameter.
Take a look at the molconverter and getatomcount user defined function examples.
A more efficient way
There is a more efficient way to evaluate user defined operators appearing in the where clause.
-
Drop all indexes (SQL: drop index idx_name) created before with the jc_idxtype indextype.
-
Execute as the JChem owner (the owner of the schema, in which JChem Cartridge has been installed):
alter indextype jc_idxtype add my_op(VARCHAR2, VARCHAR2); -
Grant execute privileges to users:
grant execute on my_func to jchemuser; grant execute on my_op to jchemuser; grant execute on jc_idxtype to jchemuser; -
Execute the following insert statement:
INSERT INTO jc_idx_udop VALUES('operator_name', 'java_class_name', 'separator', 'params_list');Description of the parameters see here .
For example:INSERT INTO jc_idx_udop VALUES('MY_OP', 'MyClass', '{SEP}', '$1{SEP}$2');(The table jc_idx_udop must currently be in the schema where the jc_idxtype index will be created, but we plan to support user defined operator in the scope of the entire JChem Cartridge installation.)
-
Implement the Java class which will evaluate the relation in the where clause. The name of the class has to be the same as the name of the previously created Java class (implements JChemCartModul.java) with the "_eval" end. For example: MyClass_eval.java
This class has to implement the chemaxon.jchem.cartridge.JChemCartEvalModule interface. The eval function of this class is called on each object returned by the JChemCartModul.java class. It returns a boolean value that represents whether the object evaluate the expression given in the SQL statement or not. The newly created Java class has to be found on the classpath of JChem Server by setting the xclasspath property in the jchem/cartridge/conf/jcart.properties file. -
Re-create your jc_idxtype indexes again. An index with the jc_idxtype indextype also has to be created on the table on which you want to use the new operator.
Take a look at the getatomcount user defined function example.
JChem-table functions
JChem Cartridge defines two procedures to perform DML operations on a table:
These procedures can be called with or without jc_idxtype index.
call jchem_table_pkg.create_jctable('myjctable', 'JChemProperties', 16, 2, 6, ', RECNO NUMBER', 'aromatize', 1);
call jchem_table_pkg.drop_jctable('myjctable', 'JChemProperties');
cdid_array := jchem_table_pkg.jc_insert('CC(N)Cc1ccccc1', 'scott.jchemtable', null, 'true', 'false');
call jchem_table_pkg.jc_update('CN1C=NC2=C1C(=O)N(C)C(=O)N2C', 'scott.jchemtable', 12, 'scott.JChemProp');
call jchem_table_pkg.jc_delete('scott.jchemtable', 'WHERE cd_id > 1000 AND cd_id < 1100', 'scott.JChemProp');
It is possible to insert more than one structure with one call. If the first parameter starts with "select" the procedure will perform the select statement and insert the resulting structures to the table. The result of the select statement should contain only one column with the structure.
cdid_array := jchem_table_pkg.jc_insert('select cd_structure from jchemtable2 where cd_id < 11', 'scott.jchemtable', null);Currently, jchem_table_pkg.jc_insert and jchem_table_pkg.jc_update can be used exclusively to manipulate JChem generated tables.
To manipulate plain (non-JChem) structure tables, the standard INSERT, UPDATE and DELETE SQL operations can be used.
Miscellaneous functions
For transformation, structure enumeration you can use jc_react, jc_transform, jc_standardize.
To convert a molecular structure into various supported text-based formats, use jc_molconvert . The result type is the same Oracle type as the original structure's. If the output structure is needed in a different Oracle type than the input structure, use jc_molconvertv , jc_molconvertb or jc_molconvertc to change it to VARCHAR2, BLOB or CLOB format respectively.
Cost estimation for the Oracle Optimizer
In order for JChem Cartridge to provide cost estimations to the Oracle Optimizer you have to perform the following steps:
You can override/modify configured defaults at both session level or operator-call level.
Enable cost estimation in JChem Cartridge
Execute the assoc_stats.sql script in the cartridge directory as the JChem-owner. For example:
sqlplus jchem/tiger@mydb @assoc_stats.sqlYou can disable cost estimation by executing the disassoc_stats.sql script in the same directory. For example:
sqlplus jchem/tiger@mydb @disassoc_stats.sqlGather system statistics
Make sure that Oracle system statistics are available to the optimizer – for example, by executing DBMS_STATS.GATHER_SYSTEM_STATS('noworkload').
Gather statistics on the structure tables
The cost estimation of JChem Cartridge relies on statistics data of certain database objects, so this must be generated prior running the calibration:
call DBMS_STATS.GATHER_SCHEMA_STATS('INDEXOWNER')INDEXOWNER is the user who created index on the structure table(s).
In order to make cost estimation be able to rely on statistics data it is essential to call statistics collection when
-
new index with jc_idxtype is created
-
if the content of the table has significantly changed
-
or the table have been re-indexed with jc_idxtype compared to what was captured during the collection.
Calibration procedure
Calibration can only be executed as the owner of the table where the structures to be searched for are stored.
-
If you want to get some details about calibration (e.g. statements used for calibration, calibrated costs) then set the following as the table owner user:
set serveroutput on; -
Run calibration as table owner. There are two possibilities:
-
Simple case
call <jchem owner>.jchem_calibration_pkg.calibrate(<tablename>, <struct. col.name>, <keyname>, <molecule>);Example:
call jchem.jchem_calibration_pkg.calibrate(’mytable’, ’cd_structure’, ’cd_id’, ’c1ccccc1’);This procedure calibrates using a select statement which combines two columns of the same table. It is worth calibrating using this procedure if in the majority of use cases combined selects are performed on the same table.
-
Combination of tables
call <jchem owner>.jchem_calibration_pkg.calibrate(<selectstatementtocalibrate>);Example:
call jchem.jchem_calibration_pkg.calibrate( ’selectcount(*)frommytable1 t, mytable2 rwherer.cd_id = t.cd_idandr.cd_id < ?andjc_compare(t.cd_structure, ’’c1ccccc1’’, ’’t:s’’) = 1’);The given select statement should contain a JC_COMPARE() call and a ‘?’ character which indicates the value to be varied during the calibration. Complex statements that combine several tables (like in the example above) are worth calibrating with this procedure.
These calls to jchem.jchem_calibration_pkg.calibrate() set the JChem costs for the actual session.
-
Permanent setup
JChem costs can be set up permanently by executing the UPDATE statement created by the calibration as JChem owner. The statement can be retrieved by the table owner from the calibration package after the calibration using the following call:
select jchem.jchem_calibration_pkg.get_update_statement from dual;This retrieval is available only in the same session where calibration was performed.
The UPDATE statement must be executed in an other session by the JChem owner.
Modifying configured default cost estimation factors
Session level overrides
The jchem_opti_pkg.set_volatile_cost_factors procedure can be used to override the default cost estimation factors for the session. It can be repeatedly called in a session changing the factors as many times as needed for subsequent calls.
The procedure accepts the following parameters:
-
opName varchar2: the identifier of the operator to which the new estimation factors are to be applied; currently the following identifiers are accepted:
-
CONTAINS
-
COMPARE
-
EQUALS
-
MATCHCOUNT
-
EVALUATE with each of them standing for to the corresponding JC_XXXX operator or jcf.XXXX function.
-
-
idxCpuCost number: the CPU cost component of the index scan estimation factor;
-
idxIoCost number: the IO cost component of the index scan estimation factor;
-
idxNetworkCost number: the network cost component of the index scan estimation factor;
-
funcCpuCost number: the CPU cost component of the function-based estimation factor;
-
funcIoCost number: the IO cost component of the function-based estimation factor;
-
funcNetworkCost number: the network cost component of the function-based estimation factor.
You can obtain the current estimation factors using the jchem_opti_pkg.get_cost_factor function which accepts the following parameters:
-
costType varchar2: accepted values are index and func
-
opName varchar2: accepted values are
-
CONTAINS
-
COMPARE
-
EQUALS
-
MATCHCOUNT
-
EVALUATE
-
-
options varchar2: the options (third argument) of the JC{_|.}COMPARE operator/function, NULL for other operators/functions.
-
resrc varchar2: the identifier of the resource, whose cost estimation component is being queried; accepted values are:
-
cpu
-
io
-
net
-
-
idxSchema varchar2 must currently be NULL;
-
idxName varchar2 must currently be NULL.
Modifying defaults for a single operator execution
The jc_compare operator accepts the following options (as part of the option list argument):
-
indexCost: <decimal-number> where <decimal-number> will be used to multiply the default cost estimation for index-scanning.
-
funcCost: <decimal-number> where <decimal-number> will be used to multiply the default cost estimation for function based execution.
Example:
In order to declare the index scan mode of substructure search with Lipinski's rule of 5s as Chemical Terms filter two times more costly than the default estimation:SELECT count(*) FROM nci_3m WHERE jc_compare(structure,'O=C1ONC(N1c2ccccc2)-c3ccccc3','sep=! t:s!ctFilter:(mass() <=500) && (logP() <=5) && (donorCount() <=5) && (acceptorCount() <=10)!indexCost:2.0') =1
Known issues
-
Parametrized similirity metrics (such as tversky) cannot be used with locales where the decimal separator is comma. (FS#9922)
-
In "user" schemata, the index type name must be qualified with the name of "index owner's" schema.
-
dbms_lob.freetemporary does not free temporary BLOBs returned by JChem Cartridge functions.
Certain functions (jcf_reactb, jcf_molconvert(b), jcf_standerdizeb, jcf_evaluateb_x) return a temporary BLOB (as the corresponding operators do). Due probably to what we believe to be an Oracle bug, when such functions are called from a PL/SQL stored procedure/function or a PL/SQL anonymous procedure, dbms_lob.freetemporary completes without an error or warning but will have no effect: the temporary BLOB created by JChem Cartridge (in a Java stored procedure) will not be freed.
Workaround:
For these functions we provide an additional parameter (temp_blob) of type BLOB, which can be used to pass to the function the locator of the BLOB which is to hold the results. If this additional parameter is set to NULL (default), JChem Cartridge will create a temporary BLOB that will be used to store the result of the conversion and this temporary BLOB will be returned to the caller. If the user assigns a BLOB locator to this additional parameter (one created by the caller using dbms_lob.createtemporary, for example), the BLOB represented by this locator will be used to store the results. In this case, the BLOB locator assigned to this additional parameter will be also returned to the caller.
Future plans
-
Pharmacophore similarity search based on ChemAxon's technologies already implemented in other tools.
-
An option to the jc_compare operator is planned to allow the specification of other set operations between the results of the individual queries (and the entire set in the structure table).
Calibration procedure (as table owner)
-
If you want to get some details about calibration (e.g. statements used for calibration, calibrated costs) then set the following as the table owner user:
set serveroutput on;
-
Run calibration as table owner. There are two possibilities:
Simple case
call <jchem owner>.jchem_calibration_pkg.calibrate(
<table name>, <struct. col. name>, <key name>, <molecule>);
Example:
call jchem.jchem_calibration_pkg.calibrate(
’CHEM_STRUCTURE_MOL’, ’MOL_FILE’, ’SRTUCTURE_FK’, ’c1ccccc1’);
This procedure calibrates using a select statement which combines two columns of the same table. It is worth calibrating using this procedure if in the majority of use cases combined selects are performed on the same table.
Combination of tables
call <jchem owner>.jchem_calibration_pkg.calibrate(<select statement to calibrate>);
Example:
call jchem.jchem_calibration_pkg.calibrate(
’select count(*) from CHEM_STRUCTURE_MOL t
join (select structure_fk
from RSLT_CALCULATED_PROPERTIES
where rownum < ?) r
on r.structure_fk = t.structure_fk
where jc_compare(t.mol_file, ’’c1ccccc1’’, ’’t:s’’) = 1’);
The given select statement should contain a JC_COMPARE() call and a ‘?’ character which indicates the value to be varied during the calibration. Complex statements that combine several tables (like in the example above) are worth calibrating with this procedure.
These calls to jchem.jchem_calibration_pkg.calibrate() set the JChem costs for the actual session.
Permanent setup
JChem costs can be set up permanently by executing the UPDATE statement created by the calibration as JChem owner. The statement can be retrieved from the calibration package after the calibration using the following call:
select jchem.jchem_calibration_pkg.get_update_statement from dual;
Note, that this action is available only in the same session where calibration was performed.