Query features JCB
Querying atoms
Organic chemists often look for molecules that cannot be represented by a single structure. Although it is possible to run multiple structure searches in cascade, it is much more efficient to run a search only once using a well designed query structure. This structure often contains query features, possibly including complex conditional expressions for atoms and bonds.
Atom lists, not lists
It is possible to define the type of an atom in a custom atom list. If the type of the corresponding atom in the target molecular structure is a member of the list, it is considered a matching atom (Table 1). Not lists can be used to specify atoms to be excluded (Table 2). Please note that the matching of not list atoms may depend on the input format of the query molecule. See details.
Table 1. Atom lists
|
|
target |
|||
|
|
|
|
||
|
query |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
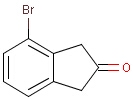
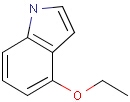
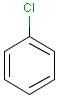


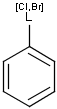
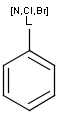
Table 2. Atom not lists
|
|
target |
|||
|
|
|
|
||
|
query |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
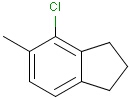
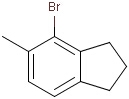
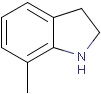
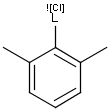
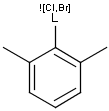
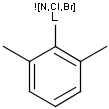
Generic query atoms
Applying atom lists and not lists is a practical solution when the number of included or excluded atoms is small. However, generic query atom types are helpful to avoid long atom lists. JChem handles at the moment the following generic query atom types. For differences between matching any atoms appearing in different file formats, see here.
A Any (any atom except hydrogen. Neither matches to explicit nor
implicit hydrogens. Please note that in JChem the SMARTS primitive "*" is imported as any atom and does not match to plain hydrogens. (Neither explicit nor implicit.)) AH Any atom, including hydrogen.
Q Hetero (any atom except hydrogen and carbon)
QH Hetero atom or hydrogen (any atom except carbon)
M Metal (contains alkali metals, alkaline earth metals, transition metals, actinides, lanthanides, poor(basic) metals, Ge, Sb and Po)
MH Metal or hydrogen
X Halogen (F,Cl,Br or I)
XH Halogen or hydrogen
Gn Member of group (column) n in the periodic system (n = 1..18)
Attention: G17 is NOT the same as X, as it contains At!
* Star atom, has the same meaning as AH query atom.
Table 3. Generic query atoms
|
|
target |
|||
|
|
|
|
||
|
query |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
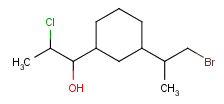
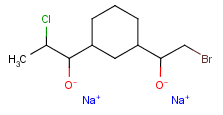
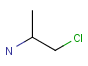
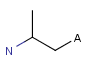
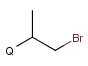
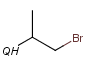
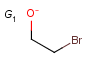
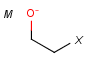
Atom properties
The chemical neighborhood of an atom is sometimes as important as its type. Conditions for the chemical environment of an atom can be defined by atom properties (Table 5). Some of these require a value (connections, smallest ring size) others don't (aromatic). The smallest set of smallest rings (SSSR) is used for the evaluation of ring properties.
a aromatic (has aromatic bond)
A aliphatic (does not have aromatic bond)
D<n> Equivalent to s<n>
H<n> total hydrogens (total number of hydrogen substituents *)
h<n> implicit hydrogens (number of implicit hydrogen substituents)
R<n> rings (number of rings the atom is a member of)
r<n> smallest ring size (size of the smallest ring the atom is a member of)
R ring membership (whether atom is part of a ring or not)
v<n> valence (total bond order)
X<n> connections (number of substituents including hydrogens)
s<n> substitution count (number of non-hydrogen substituents
plus number of isotopic hydrogen substituents)
s0-s5: exact substitution count; s6: 6 or more substitutions
s* substitution as drawn (no extra non-hydrogen and no extra isotopic hydrogens)
rb<n> ring bond count (number of ring bonds next to the atom)
rb0, rb2-rb3: exact ring bond count; rb4: 4 or more ring bonds.
The same property can be achieved using the SMARTS "x" property (see smarts doc).
rb* ring bond count as drawn (no extra ring bonds)
u unsaturated atom (atom has double, triple or aromatic bond)
* Corresponds to both MDL and Daylight behaviors, depending on the source of the Molecule object. For details, see the differences section.
Table 4. Atom properties
|
|
target |
|||
|
|
|
|
||
|
query |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
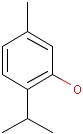
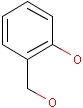
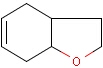
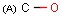
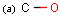
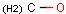
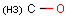
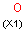
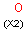
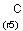
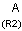
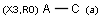
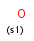
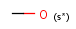
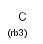
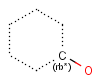
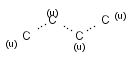
Isotopes, charges, radicals
Isotopes, charges and radicals are atom properties which can be found not only on the query structure, but on a target as well. They are considered during the search. This includes that an atom without isotope, charge and radical information will match to atoms with information about those properties. However, this behavior can be fine-tuned using the following options:
-
setOption(OPTION_CHARGE_MATCHING, CHARGE_MATCHING_DEFAULT / CHARGE_MATCHING_EXACT / CHARGE_MATCHING_IGNORE)
(Cartridge: charge:d/e/i jcsearch: --charge:d/e/i) -
setOption(OPTION_ISOTOPE_MATCHING, ISOTOPE_MATCHING_DEFAULT / ISOTOPE_MATCHING_EXACT / ISOTOPE_MATCHING_IGNORE)
(Cartridge: isotope:d/e/i jcsearch: --isotope:d/e/i) -
setOption(OPTION_RADICAL_MATCHING, RADICAL_MATCHING_DEFAULT / RADICAL_MATCHING_EXACT / RADICAL_MATCHING_IGNORE)
(Cartridge: radical:d/e/i jcsearch: --radical:d/e/i)
(All constants above are defined in class chemaxon.sss.SearchConstants.)
The following tables show some examples.
Table 5.
|
|
target |
||
|
|
|
||
|
setOption(OPTION_CHARGE_MATCHING, CHARGE_MATCHING_DEFAULT) (Default) |
|||
|
query |
|
|
|
|
|
|
|
|
|
setOption(OPTION_CHARGE_MATCHING, CHARGE_MATCHING_EXACT) |
|||
|
query |
|
|
|
|
|
|
|
|
|
setOption(OPTION_CHARGE_MATCHING, CHARGE_MATCHING_IGNORE) |
|||
|
query |
|
|
|
|
|
|
|
|
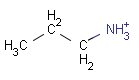
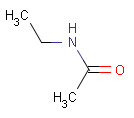


Table 6.
|
|
target |
||
|
|
|
||
|
setOption(OPTION_ISOTOPE_MATCHING, ISOTOPE_MATCHING_DEFAULT) (Default) |
|||
|
query |
|
|
|
|
|
|
|
|
|
setOption(OPTION_ISOTOPE_MATCHING, ISOTOPE_MATCHING_EXACT) |
|||
|
query |
|
|
|
|
|
|
|
|
|
setOption(OPTION_ISOTOPE_MATCHING, ISOTOPE_MATCHING_IGNORE) |
|||
|
query |
|
|
|
|
|
|
|
|
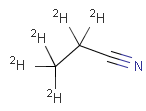
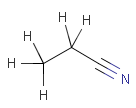
Table 7.
|
|
target |
||
|
|
|
||
|
setOption(OPTION_RADICAL_MATCHING, RADICAL_MATCHING_DEFAULT) (Default) |
|||
|
query |
|
|
|
|
|
|
|
|
|
setOption(OPTION_RADICAL_MATCHING, RADICAL_MATCHING_EXACT) |
|||
|
query |
|
|
|
|
|
|
|
|
|
setOption(OPTION_RADICAL_MATCHING, RADICAL_MATCHING_IGNORE) |
|||
|
query |
|
|
|
|
|
|
|
|
Link nodes (link atoms)
Link nodes are atoms which may occur one or more times defining a variable length chain or ring. The link node is denoted by its brackets and the repetition range. All bonds not crossed by the brackets (and connecting parts) are also repeated together with the link node. See examples below.
Table 8.
|
Query |
Possible meanings |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
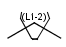
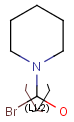
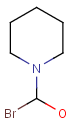
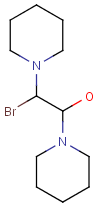
Repeating units
Repeating units represent structural parts that can be repeated several times. The repeating unit is enclosed in brackets with one or two head and the same number of tail crossing bonds. (Head crossing bonds go through the left bracket.) Two bond pairs represent ladder type repeating units. The repetition range is a comma-separated list of possible repetitions or repetition intervals, e.g. "1,3,5-9". The repetition pattern specifies the way how the subsequent repeated units are linked together: it can be head-to-head(hh), head-to-tail(ht) or either/unknown(eu) (the either/unknown case is not handled by the search software). In case of ladder type polymers there is also a flip(f) option that defines that the top and bottom crossing bonds are flipped during each connection.
Table 9.
|
Query |
Subscript explanation |
Possible meaning |
|
|
|
Unit repeats 1 or 3 times with head-to-head connectivity. |
|
|
|
|
Unit repeats 2 to 3 times with head-to-tail connectivity, no flip. |
|
|
|
|
Unit repeats 2 to 3 times with head-to-head connectivity and flip. |
|
|
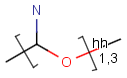
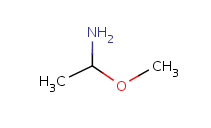
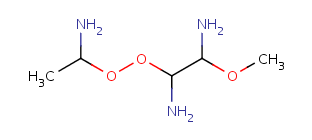
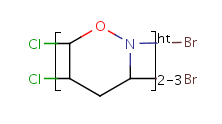
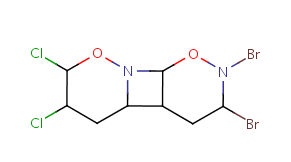
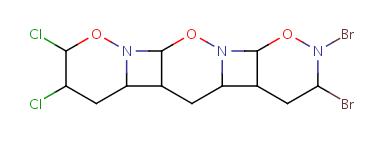
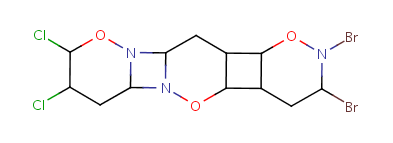
Table 10. Substructure search examples
|
Query |
Target |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Undefined R-atoms
An undefined R-atom is an R-group without definitions. (For the description of defined R-atoms, see R-group query structures .)
By default, undefined R-atoms can match atom groups including one or more heavy atoms or a hydrogen isotope. Hydrogen isotopes are handled as non-hydrogen atoms: they are forbidden at blocked positions and considered as heavy atoms when matching undefined R-atoms.
The matching behavior of undefined R-atoms can be altered using the undefinedRAtom option:
g Default: Undefined R-atom matches a group of
one or more connected atoms in target,
including at least one heavy atom.
gh Undefined R-atom matches a group of
one or more connected atoms in target,
which can also be a single H atom.
ghe Undefined R-atom matches a group of
one or more connected atoms in target,
which can also be a single H atom or the empty set
(empty set match is allowed for isolated or
one-attachment R-atoms only).
a Undefined R-atom matches any single atom in target.
u Undefined R-atom matches only an undefined R atom in target.
Notes:
-
g, gh, and ghe options are not supported in case of R-group queries of Markush targets.
-
If group matching is switched on (options g, gh, or ghe) and at least one undefined R-atom is present in the query, then substitution is blocked at all other positions. In other words, this means that a full fragment matching is performed even if otherwise substructure search was specified. (Note, that in case when there is no undefined R-atom in the query, individual positions can be blocked using the s* query property.)
Table 11/A. Examples with default settings
|
Query |
Target |
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
There are two more options which affect the R-atom group matching behavior:
-
bridgingRAllowed (n/y): if switched on (y) then different R-atoms are allowed to match the same group of atoms (default: switched off (n)).
-
RLigandEqualityCheck (y/n): if switched on (y) then R-atoms with the same R-group ID should match ligands with the same structure (default: switched on (y)).
Note, that bridging R-atom matches are also ligands having the same structure.
Table 11/B. Examples with specific options
|
Query |
Target |
|||||
|
|
|
|
|
|
|
|
|
bridgingRAllowed:y |
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
undefinedRAtom:ghe, RLigandEqualityCheck:n, bridgingRAllowed:n |
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
When undefined R-atoms matching is group matching and hydrogen with or without empty set matching is also allowed (options gh, ghe ), then for each undefined R-atom the possible matching atom groups are sorted by the following order of precedence:
-
group or isotope hydrogen
-
explicit hydrogen
-
implicit hydrogen
-
the empty set
This ordering is done locally when a scaffold matching is extended by group matchings in place of the undefined R-atoms.
However, if the --hitOrdering:g parameter is set, then this ordering is global over all search hits, in the order of R-atom numbers (R1 atom matches are considered first, then R2 matches, etc.). For example, if there are R1, R2 and R3 R-groups then the sorting algorithm will try to match R1 to a heavy group if possible, otherwise a H-atom and finally to the empty set; then the same is played with R2 and then with R3.
Limitations when undefined R-atoms are matching group (options g, gh, ghe ):
-
The query is not allowed to have two adjacent undefined R-atoms or an undefined R-atom adjacent to a homology group.
-
Undefined R-atoms adjacent to H atoms only are not allowed.
-
Query features on undefined R-atoms are not allowed.
-
Double bond stereo involving undefined R-atom is not allowed.
-
Undefined R-atom as chiral center is not supported, but it can be adjacent to chiral centers. Examples:
not supported
supported
-
The "rb*" query property is ignored on all atoms in the query.
-
SMARTS atom / bond properties referring to the query may not be processed correctly.
-
Search is always processed in order sensitive mode.
SMARTS atoms
JChem's search supports all valid SMARTS atom expressions. (See Daylight's SMARTS theory manual.)
SMARTS atoms are depicted the following way in Marvin:
The following additional query features are handled as part of this:
Logical operators between query elements
This query feature allows the use of logical operators: the two "and" operators, "or" and "not" to combine queries into complex expressions. Table 12 shows the operators in the order of their precedence ("!" evaluated first):
Table 12.
|
Operator |
Name |
|
! |
not (unary operator) |
|
& |
high precedence and (default operator, i.e. can be omitted between two query expressions) |
|
, |
or |
|
; |
low precedence and |
Table 13. Examples
|
Query |
Target |
|||
|
NCC(O)=O |
[N+]CC([O-])=O |
[H]OC(=O)N([H])C |
COC |
|
|
[OX2H,OX1-] |
|
|
|
|
|
[O&X2&H,O&X1&-] |
|
|
|
|
|
[NX3;H2,H1] |
|
|
|
|
|
[OX2!-] |
|
|
|
|
Recursive SMARTS
One of the most powerful feature of SMARTS atoms is recursive SMARTS. It can be used to describe an environment of an atom with the syntax "$( <<SMARTS expression>> )". The first atom of the <<SMARTS expression>> will be matched to the atom in question, and the rest to its environments. It evaluates true if the expression matches.
Table 14.
|
SMARTS |
Meaning |
|
[OX2$(OaaN)] |
Aliphatic oxygen with two connections, next to an aromatic ring having an aliphatic N in ortho position. |
|
[OX2$(*aaN)] |
Same as above. |
|
[$([OX2]aaN)] |
Same as above. |
|
[NX3;H2,H1;!$(NC=O)] |
Primary or secondary amine, not amide. |
|
[$(N~*~*~[O!$(O([C,c])[C,c])])] |
Aliphatic N three bonds away to a non-ether aliphatic O. |
Table 15.
|
Query |
Target |
|||
|
|
|
|
||
|
[OX2$(OaaN)] |
|
|
|
|
|
[$(OCC),$(OCN)] |
|
|
|
|
|
[$(O([C,c])[C,c])] |
|
|
|
|
|
[$(N~*~*~[O!$(O([C,c])[C,c])])] |
|
|
|
|
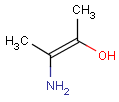
Please note that uppercase atom symbols only match to aliphatic atoms and lowercase only to aromatic.
Further information regarding SMARTS
In JChem explicit and implicit hydrogens in the target are treated the same, and hence the presence or absence of plain hydrogens does not affect the result of the search.
In JChem the SMARTS primitive "*" (any atom) does not match to plain hydrogens. (Neither explicit nor implicit.) However, it matches deuterium and charged H. See details.
Further SMARTS examples can be found on Daylight's page.
Pseudo atoms
Pseudo atoms have user-defined atom types, and they only match another pseudo atom of the same name (case insensitive). Commonly used pseudo atoms include "Resin" and "Pol", referring to the often used solid phases in syntheses (Pol is the default pseudo for resin in MDL ISIS/Draw).
Table 16.
|
Query |
Target |
|||
|
|
|
|||
|
|
|
|
||
|
|
|
|
||
|
|
|
|
||
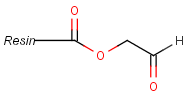
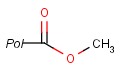
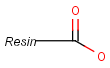
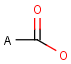
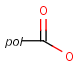
It should be noted that there is no chemical intelligence associated with pseudo atoms. This means that if a common abbreviation is used as pseudo atom, it will not match the corresponding molecular group. To achieve this, correct abbreviations (Superatom S-groups) must be used.
Homology (generic) groups
Some pseudo atoms represent homology groups, which refer to specific chemical classes. Examples are alkyl, heteroaryl, protecting, etc. (see complete list) From JChem version 5.3 homology groups are supported on the query side. Target side handling was already implemented in version 5.1. Query side homology searching uses the same homology definitions as the target side except a few cases. Query side homology handling does not require any special licenses beside JChemBase or changing search options from the default behavior.
Homology matching
In case of query-side homology handling the two main classes of homology groups are dealt separately. These are the built-in groups and the user_defined groups and their description can be found here.
Built-in groups
Built-in groups match on target structures that fulfill the requirements of the given structure. Some examples are shown below with differently colored part matched by the homology groups.
Table 17. Searching with built-in homology groups
|
Query |
Target |
Hit |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
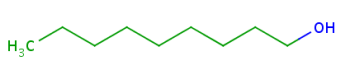
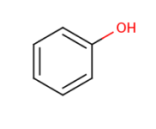
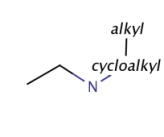
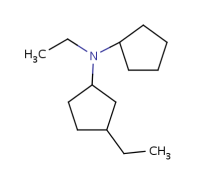
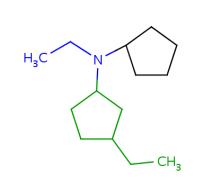
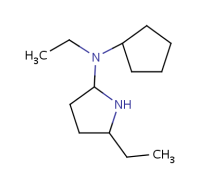
About the retrieved hit indexes see the appropriate section.
User-defined groups
User-defined homology groups are transformed into an R-group representation using their R-group definitions. These are used for searching with the r-group searching functionality. The R-group definitions are customizable and the addition of new groups is also possible.
According to the features supported by Rgroup search the following limitations apply:
-
Only the index of the target atom matched by the first atom of the Rgroup definition is retrieved not all the target indexes belonging to the homology group.
-
Hit variations where only the index of the homology atom changes are not retrieved.
Which atoms will be included in a homology substructure?
User-defined groups
The matching of the previously described user-defined homologies is straightforward based on R-group searching. One of the definitions must be fully present in the target structure. If allHits option is chosen then because of the operation of R-group searching hits differing only in the matching of the R atom (user-defined homology) are not retrieved.
Built-in groups
Built-in groups always match such a portion of the target structure, that is in accordance with the homology group's definition rules.
Such portions are extended with optional parts (see definitions) as much as possible. Table 18. shows an example, the alkenyl atom has to match on atoms 5-6 and this is extended by the remaining part of the alkyl chain (atoms 1-4).
Table 18.Hit of a built in group
|
Query |
Hit |
|
|
|
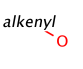
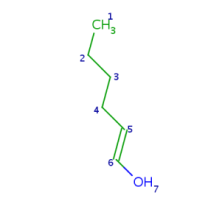
Retrieving the largest possible substructure
To avoid the combinatorial explosion of the algorithm and all substructure results, homology atoms match in a greedy manner. This means that only the largest possible substructure matches are returned, partial substructures are not. See example in Table 19.
Table 19."Illustration for all hit retrieval rules: only the largest possible substructure is returned.
|
Query |
Hits |
Not retrieved |
|
|
|
|
|
|
|
By expanding the homology group's hit it is ensured that every atom that may be matched by the group will be matched.
Matching on homology atoms
Searching homology groups on homology groups is possible under the limitation described below. However overlapping matching of homology groups on homology groups and specific parts is not supported (e.g. alkyl=C does not match on alkenyl and vice versa).
Specific options and limitations
Some search options and features have restricted support with query side homology groups:
completeHG option
This option is meaning full for target homology groups. Setting to false meant that in case of substructure searching the query can be a subset of what would be a complete homology group (see completeHG option). When homology group is on the query side then it must have a full matching structure on the target side even in case of substructure search.
query properties
Query properties on homology atoms have no effect, but they can be used in the usual way on other atoms of the same query structure. The only exceptions are the sand s* properties, which are considered for query side homology atoms as well.
polymers
Polymers can be part of the same query structure, but homology groups inside polymers are not supported.
markush targets
Query side homology groups are not supported with target markush structures (markush searching) if query side homology handling is enabled (narrow translation is on). When searching with query side homology groups the only allowed markush feature are the built-in homology groups.
Query-side homology handling (homology narrow translation) option
The homology narrow translation option can switch off special handling of homology groups, hence they can match the homology group of the same type only (e.g. alkyl on alkyl or chk). More information about translation options can be found in the markush search documentation. The default value for homology translation is NONE (translation is not enable on any query atom).
Rgroups with more than two attachment points
Rgroups with more than two attachment points are not supported on the query side.
location in ring
Only the following homology groups are applicable in rings: atomic homolgy groups (AnyAtom, Metal, AlkaliMetal, OtherMetal, TransitionMetal, Lanthanide, Actinide) and RingSegment.
UnknownGroup
UnknownGroup homology group in query structure is matched only with UnknownGroup homology groups in target structure.
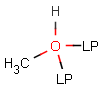
Lone pairs
JChem search can handle query and target atoms having lone pairs associated with them. Lone pairs on the query side match explicit and implied lone pairs, but please note that lone pairs are only considered when attached to an atom, i.e. isolated lone pairs will not match anything.
Table 20.
|
Query |
Target |
|||
|
|
|
|
||
|
|
|
|
|
|
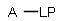
Querying bonds
Generic bonds
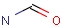
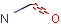
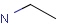
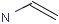
Querying against bonds can determine if a bond in the target molecule is one of the four basic types (single, double, triple, aromatic) or one of the generic types that are available for fine tuning query structures (Table 6). The line style represents the type of a bond.
|
|
any |
|
|
single or double |
|
|
single or aromatic |
|
|
double or aromatic |

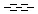

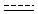
Table 21. Generic bond types
|
|
Target |
|
|
|
||
|
Query |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
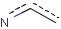
Note on aromatic bonds
For the correct use of aromatic bonds and aromatic systems in general, see the Aromatization section under Standardization .
Stereo bonds (tetrahedral chirality and cis/trans configuration)
See section Stereochemistry.
Chain/ring bond attributes
In addition to the bond type discussed above, a bond topology query attribute can be assigned to bonds. This expresses that the bond must be part of a ring or must not. See the examples below.
Table 22. Generic bond types
|
|
Target |
||
|
|
|
||
|
Query |
|
|
|
|
|
|
|
|
|
|
|
|
|


SMARTS bonds
SMARTS bond expressions are also supported. (See Daylight's SMARTS theory manual.)
SMARTS bonds are depicted the following way in Marvin:
Like at SMARTS atoms, SMARTS logical operators "!" (not), "&", ";" (high and low precedence and), "," (or) can be used. "&" is the default operator, hence "and" is assumed if there is no operator between two SMARTS primitives. Furthermore, the following characters have valid meanings:
Table 23.
|
Bond expression |
Meaning |
|
- |
Single bond |
|
= |
double bond |
|
# |
triple bond |
|
: |
aromatic bond |
|
@ |
any ring bond |
|
/ |
directional bond: single "up" (used at cis/trans) |
|
\ |
directional bond: single "down" (used at cis/trans) |
Table 24.
|
SMARTS |
Meaning |
|
C-,=,#C |
Two aliphatic carbons connected by single, double or triple bond. |
|
*-!@* |
Two atoms connected by a nonring single bond. |
|
*@-,!@&/*=*@-,!@&/* |
Double bond between two single bonds in ring or not in ring but in trans configuration. |
Table 25.
|
Query |
Target |
|||
|
|
|
|
||
|
C-,=,#C |
|
|
|
|
|
*-!@* |
|
|
|
|
|
*@-,!@&/*=*@-,!@&/* |
|
|
|
|
Further SMARTS examples can be found on Daylight's page.
Coordinate bonds
Coordination compounds can be registered and searched for in JChem structure databases. Both "atom to atom" and "multicenter" (involving more than two atoms) representations are supported.
Atom to atom coordinate bonds
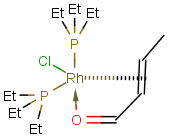
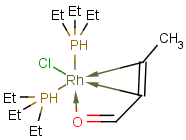
Matching of "atom to atom" coordinate bonds is similar to matching other bond types. The direction of the coordinate bond arrow is not checked. See examples below. (Q stands for hetero atom, M for any metal atom. The thin dotted bond represents an ANY query bond.)
Table 26.
|
Query |
Target |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
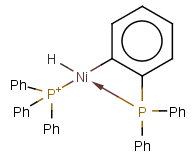
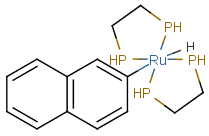



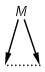
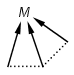
Multicenter coordinate bonds
Multicenter coordinate bonds are handled the way as if each atom at opposite ends of the coordinate bond had individual coordinate bonds in between them. This means that the following molecule pairs are equivalent (The used molecule representation conforms to IUPAC recommendation: atom to atom coordinate bonds are displayed by an arrow and multicenter coordinate bonds are denoted by thick dashed line.)
|
|
|
|
|
|
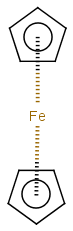
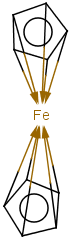
So individual and multicenter representations can both be used during searching, in all combinations. See examples below. (The thin dotted bonds represent ANY query bond types.)
Table 27.
|
Query |
Target |
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|

Position variation bonds
Position variation bond (or variable point of attachment) is used to express that a bond may be attached to multiple positions (atoms), most often used for rings. This is represented by a multicenter atom at one or both end of the position variation bond. Its representation and drawing is described in the Marvin Sketch help and in Marvin JS help . See examples below.
Table 28. Meaning of position variation.
|
Query |
Possible meanings |
|
|
|
|
|
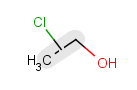
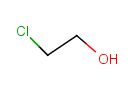
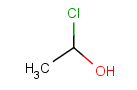
Table 29. Matching of position variation queries.
|
Query |
Target |
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
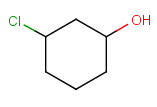
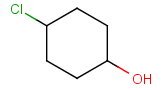
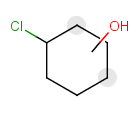
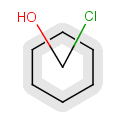
Components
This section describes the features related to different types of components. Component = set of connected atoms in a molecular drawing. The connection can be:
-
Connection by bonds
-
A component bracket (S-group)
-
A reaction component
The handling of these different types of components is described below.
SMARTS component level grouping
This feature uses components as atoms connected by bonds. In SMARTS queries it can be specified whether different components (fragments) of the query should appear in the same or different components in the target. It is represented by grouping parentheses around the component in the SMARTS string. Please note that there is no graphical representation of this feature in Marvin.
Table 30.
|
SMARTS representation |
Meaning |
|
C.C |
No restrictions. |
|
(C.C) |
The two carbons must appear in the same component. |
|
(C).(C) |
The two carbons must appear in different components. |
Table 31.
|
Query |
Target |
|||
|
|
|
|
||
|
C.C |
|
|
|
|
|
(C.C) |
|
|
|
|
|
(C).(C) |
|
|
|
|
|
(C).(C).C |
|
|
|
|
Component, Mixture and Formulation brackets
This feature relates to the use of brackets (S-groups) of type COM (component), FOR (formulation) and MIX (mixture). A component here is a set of atoms contained by a component bracket.
Ordered and unordered mixtures
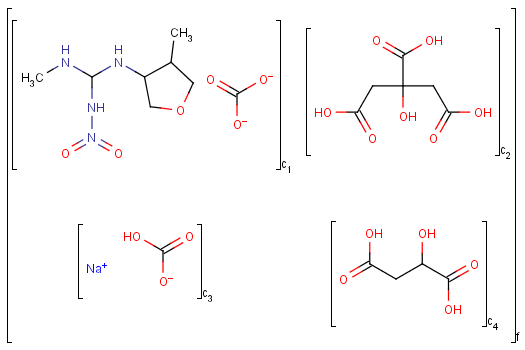
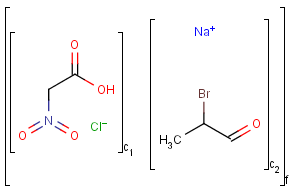
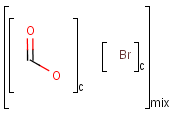
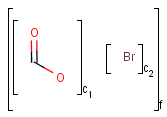
An unordered mixture (MIX type S-group) consists of several unordered components. For these types of mixtures, the order of addition during the preparation is not important. Example:
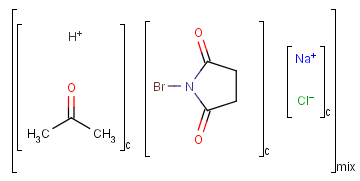
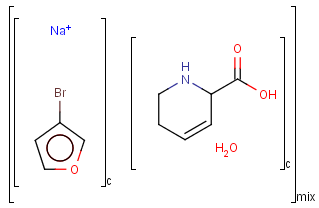
Ordered mixtures (FOR type S-groups), on the other hand contain ordered components, which define the order of addition. Example:
Matching of mixture and component brackets
The component grouping of component brackets is considered during the matching, so all atoms drawn inside component brackets in the query can only match atoms that are contained in the same component brackets in the target and separate components can only match separate components.
Component brackets without surrounding mix or for brackets are considered as being in mix (unordered mixture) brackets and molecules not drawn in any component brackets are considered to be in the same component.
Table 32.
|
Query |
Target |
|||
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|


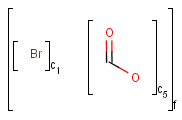
Matching of formulation brackets
Unordered mixture (mix) queries match both unordered (mix) and ordered (for) mixtures. However, ordered (for) mixtures only match ordered mixtures, and the component numbering must keep order. Examples:
Table 33.
|
Query |
Target |
||
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Other component features
During reaction searching, reaction component grouping is maintained; see at the reaction component handling section.
Full fragment matching ensures that all query components (atoms connected by bonds) match only full components. See its description in the Search types section.
Explicit hydrogens
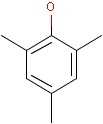
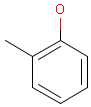
For the sake of simplicity, organic chemists usually do not draw hydrogen atoms on molecules, but in some models used to represent molecules the hydrogens are shown implicitly or explicitly. Whatever display mode one prefers, all free valences of the atoms are considered to be filled with hydrogens. In case of query structures, explicit query hydrogens have a significant importance. An explicitly drawn query hydrogen defines that the target must contain a hydrogen in that position (Table 34).
Table 34.
|
|
Target |
|||
|
|
|
|
||
|
Query |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
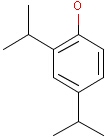
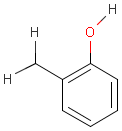
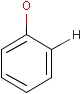
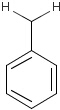
Searching with empty structures
Because of its special behavior, we separately discuss the use of empty queries. For different search types, the results of empty structure search can be seen in Table 35:
Table 35.
|
Search type |
Empty query retrieves |
|
|
memory search |
database search |
|
|
substructure |
all structures |
|
|
full fragment |
all structures |
|
|
full |
empty structures |
|
|
duplicate |
no hits |
|
|
superstructure |
empty structures |
empty structures |
In case of duplicate and superstructure search types, the results might need explanation:
-
Duplicate search doesn't retrieve any hits because in many applications the empty molecule represents an unknown structure. To enable inserting more than one empty structures in a database table with duplicate filtering, duplicate search gives no hit.
-
In most cases superstructure search retrieves only empty structures as expected. There is a different behavior for query tables only. Empty query substructure searches conventionally retrieve all records of a table. As substructure search is not allowed for query tables, its default superstructure search with an empty query can be used to retrieve all entries.
Furthermore, if the query structure is non-empty, but the target molecule is empty, then neither of the search types listed above retrieves hit (both in case of memory and database search). In case of superstructure search, this behavior is not necessarily intuitive, but its reason is that empty structure typically represents an unknown compound.
Chemical Terms filtering expressions
Searches can include extra conditions formulated in the Chemical Terms language. Chemical Terms is a chemistry language which allows users to formulate complex chemical questions, expressions and rules. Chemical Terms can contain references to functional groups, other structural elements and physico-chemical properties. The syntax is described in the Chemical Terms Reference. Search specific functions contained in the search context provide access to the query and the target molecules, the search hit array and its elements:
-
mol(), target(): both refer to the search target molecule
-
query(): refers to the search query molecule
-
m(int i): refers to the query atom index with atom map i
-
hit(), h(): both refer to the search hit array
-
hit(int i), h(int i): both refer to the i-th element of the search hit array, this is the target atom index matching the query atom with atom index i
-
hm(int i): refers to the target atom index matching the query atom with atom map i (shorthand for h(m(i)))
The default input molecule is the target molecule (e.g. mass() is the same as mass(target()), both refer to the molecule mass of the target molecule).
The filtering expression can be set by
-
setChemTermsFilter(value)
-
setChemTermsFilterConfig(value)
in the SearchOptions class. The latter specifies a configuration XML with function/plugin definitions to be used in addition to those provided by the built-in evaluator.xml .
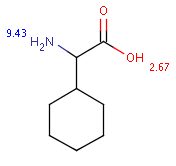
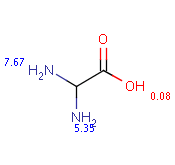
The following table shows some examples (pK a values are shown at target atoms).
Table 36.
|
|
Target |
||
|
|
|
||
|
setFilter("pka(hm(1))> 2") |
|||
|
Query |
|
|
|
|
|
|
|
|
|
setFilter("pka('acidic', hm(1))> 2 && mass()> 100") |
|||
|
Query |
|
|
|
|
|
|
|
|
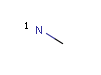
A set of working examples is also available.
Attached data
Attached data is a custom field assigned to atoms or brackets. It has an identifier string (name) and a value. Furthermore, a query operator can describe different restrictions in queries.
By default attached data is ignored in search (except in case of duplicate search - see Attached data in duplicate search). When matching is enabled (by the attachedDataMatch search option), attached data fields in the query and target with the same name are compared. If only a subset of the attached data fields should be compared, the names of data tags required for comparison can be set using the attachedDataPrefixes option. Two matching modes are available to compare a query-target data field pair:
-
In general mode the value in target molecule must
-
satisfy the condition of query molecule (when there is an operator defined in query)
-
be equal to the value of query molecule (when there is no operator defined in query)
-
-
In exact mode both operators and values must match. An attached data existing only in target prevents matching.
Operators available are: <, >, =, <=, >=, <>, BETWEEN, LIKE, CONTAINS
Remarks
-
When using the BETWEEN operator, value must contain two numbers - separated by space(s) -, the inclusive borders of the interval.
-
In case of LIKE operator wildcard characters "%" and "_" can be used. "%" stands for at least one unspecified character and "_" stands for exactly one unspecified character. To enter patterns that contain "%" or "_" characters as non-wildcards, use the symbols "\%" and "\_". Matching is case-insensitive.
Table 37. Search examples using attached data matching in general mode. (The structure diagrams usually display the value and query operator only. But if the pointer is moved over an attached data field in Marvin Sketch, the field name and the related atoms are also displayed as part of the colored highlight. This depiction is used below, to show all required information to understand the matching behavior.)
|
Query |
Target |
|
|
|
|
|
|
|
|
|
|
|
|
|
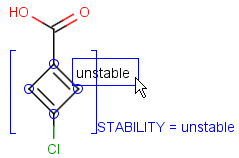
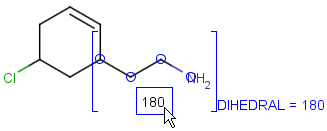
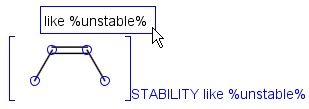

Table 38. Field matching examples
|
Attached data on query molecule |
Attached data on target molecule |
Matching mode |
|||
|
operator |
value |
operator |
value |
exact |
general |
|
|
9 |
|
9 |
|
|
|
= |
9 |
|
9 |
|
|
|
BETWEEN |
7 12 |
|
9 |
|
|
|
BETWEEN |
7 12 |
BETWEEN |
7 12 |
|
|
|
LIKE |
%but_ne |
|
2-Butane |
|
|
Attached data in duplicate search
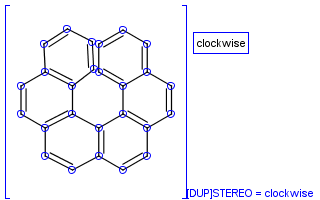
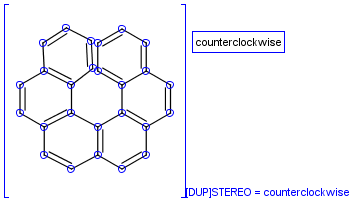
To help represent new molecular features during duplication control, from JChem 5.3 duplicate search uses special attached data with name starting with "[DUP]". These attached data names are automatically checked during duplicate searches (in exact mode).
Table 39. Example: Special attached data checking during duplicate search (differentiating enantiomers of helicene)
"Helicenes are notable for having chirality while lacking both asymmetric carbons and chiral centers.
Helicenes' chirality results from the fact that clockwise and counterclockwise helices are non-superimposable." ( Wikipedia )
|
Query |
Target |
|
|
|
|
|
|
|
|
|
|
|
|
|