Pharmacophore Fingerprint PF
This manual gives you a walk-through on how our pharmacophore fingerprint implementation works:
Pharmacophore features
Pharmacophoric features include all those binding related structural or chemical properties of chemical compounds that are thought to be responsible for a specific pharmacological action. Chemical features taken into account usually include hydrogen bond donor/acceptor, charge, hydrophobicity and aromacity. These features (or pharmacophoric properties) are arbitrary in that sense that users of the software can select the pharmacophoric features to be considered and can even provide the custom definition of the particular feature (for example specify under which circumstances is an atom aromatic). In the present approach individual atoms are labeled with pharmacophore properties. These labeled atoms are often referred to as pharmacophore points.
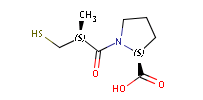
Fig. 1 Captopril, a drug that reduces peripheral arterial resistance
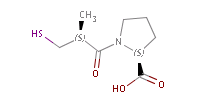
Fig. 2 Pharmacophore points in captopril: all three oxygen atoms act as H bond, while the S is both an acceptor and a donor (indicated ba colours)
There are numerous methods available to perceive pharmacophore points in chemical compounds. One family consists of methods that are based on calculation (quantitative approximation) of chemical properties (for example pKa, electronic potential). Another big family is the knowledge- or rule-based approaches. ChemAxon's pharmacophore point identification tool uses a custom expression evaluator module enabling any of these methods and even their combination. The pharmacophore definitions are provided in an XML configuration file.
The basic observation that makes rule based methods feasible is that the pharmacophore character of an atom is highly determined by the atom itself and its local neighborhood. The mainly local nature of pharmacophore characteristic of atoms enables the use of structural patterns to capture a certain local arrangement of atoms (in an arbitrary larger molecular environment).
For example consider primary amides:

The oxygen atom shows hydrogen bond acceptor characteristic, while the nitrogen atom exhibits hydrogen bond donor characteristic independent from the rest of the molecule.
However, the situation is not always that simple. Chemistry is an infinite source of exceptions to such simple rules, not surprisingly there are always exceptions to generic rules in the case of pharmacophore characterization too. Proper handling of such exceptions needs the specialization of rules, which at the end necessitates the use of logical (or boolean) expressions in constructing rules. For example a viable rule that holds in most cases is that nitrogen in tertiary amines is hydrogen bond donor. However, there are exceptions to this rule, thus it has to be extended: nitrogen in tertiary amines is hydrogen bond donor, except when attached to an sp2 atom or, for instance, when part of N-cyano-methyl piperidine.
Fig. 3 Tertiary amine, N is protonated at pH 7.0 and it is a H bond donor.

Fig. 4 In the case of tertiary amine where the N is next to an sp2 atom, its lone electron pair delocalizes, thus it is neither a donor nor an acceptor

Fig. 5 N-cyano-methyl piperidine is another exception to the general rule: the N in the piperidine ring is deprotonated at pH 7.0, so it is neither a donor, nor an acceptor.
At the end of the pharmacophore point type perception process the chemical graph of the molecule to be perceived is labeled with pharmacophoric features. This information can be visualized in MarvinView using pharmacophore type dependent coloring or simply be printed as a pharmacophore point list, which can also be stored in MDL SDfiles.
The following table gives an example: the compounds are given as SMILES strings and their pharmacophore maps are generated. Each pharmacophoric feature is denoted by one character: h: hydrophobic, a: hydrogen bond acceptor, d: hydrogen bond donor, r: aromatic, +: cationic character, -: anionic character. In the case when a pharmacophoric point has mixed characteristic, feature symbols are separated by a slash character.
|
SMILES |
name |
pharmacophore map |
|
CCC=O |
1-propanal |
h;h;h;a |
|
CCCO |
1-propanol |
h;h;h;a/d |
|
C1=CC=CC=C1C(C)Cl |
1-chloro-ethyl-benzene |
r;r;r;r;r;r;h;h;h |
|
SC(CN)C(O)=O |
cystein |
d;h;h;+/d;-;-/a;-/a |
Table 1. Some example compounds with their names and the generated pharmacophore maps
Pharmacophore fingerprints
Pharmacophore fingerprints attempt to model binding related structural or chemical properties of chemical compounds with the use of simple statistics of chemical features. In the case of pharmacophore fingerprints generated these features are always assigned to individual atoms of the molecule thus these fingerprints are atom based pharmacophore fingerprints.
Another characteristic of the particular pharmacophore fingerprints used in our software is their two-dimensional (2D) nature. Although, the widely used term, 2D pharmacophore fingerprint is misleading as fingerprints themselves are inherently one-dimensional constructs. Not fingerprints, but molecular structures are, within this context, considered as being two-dimensional: spatial position of atoms is either not known or is neglected in fingerprint generation.
Pharmacophore models assemble the set of pharmacophore points along with their relative arrangement, which, in the simplest case, is the three-dimensional Euclidean distance between each point pair. However, in the two-dimensional case no spatial information is available, thus topological relations are used to represent the relative position of pharmacophore points. The most apparent counterpart of Euclidean distance in the three dimensional space is the topological distance in the topological space of the chemical graphs. This distance is equal to the length of the shortest path between two nodes (atoms) of the chemical graph, that is the smallest number of graph edges (bonds) connecting the two atoms.
A further choice in constructing pharmacophore models is how these relative positions are built into the model. Common approaches consider either all pharmacophore point pairs or all pharmacophore point triplets (triangles). In the approach taken by GenerateMD only pharmacophore pairs are used.
The example below shows all pharmacophore feature pairs and the corresponding shortest paths for the captopril:
|
|
|
|
|
|
|
|






Fig. 6 All pharmacophore feature pairs and the shortest paths between them for captopril
Atom-pair based 2D pharmacophore fingerprints are defined as the collection of all atom-atom pharmacophore feature pairs along with their topological distances. In order to make the handling of this often large amount of data easier, the data set is represented in histograms. One histogram is associated with each pharmacophore feature pair (for example acceptor-acceptor, donor-positive, negative-hydrophobic etc.), thus the total number of histograms is f(f+1)/2, where f denotes the number of pharmacophore features used.
These histograms have the same number of bins. Bins are associated with the distances between pharmacophore points. Since topological distances are discrete values one bin belongs to one certain distance value. The bin labeled with d stores the number of pharmacophore point pairs (of the corresponding type) that are d bonds apart from each other. The distance range of the histogram can be set by specifying a minimal (m) and maximal (M) distance. Values below minimum are put in the first bin (i.e. the one labeled with m), while those greater than the maximum are put in the last one (labeled with M). The pharmacophore fingerprint is the ordered sequence of the feature pair distance histograms.
|
|
|


Fig. 7 Pharmacophore feature pair histograms and the pharmacophore fingerprint of captopril
-
Pharmacophore features are not necessarily directly assigned to atoms, for instance, the center of a benzene ring rather than its individual atoms can be characterized as aromatic. However, there is no evidence found in the relevant literature that such non-atom-based approaches have any advantages over atom-based descriptions.
-
For the sake of simplicity only hydrogen bond donor and acceptor properties were used in the example. Apparently, there are no limitations to either the number or the kind of pharmacophore types used in the pharmacophore fingerprints.
-
Global effects that involve the entire molecule are also known, for instance tautomerism gives an apparent example of such mechanisms.
Pharmacophore fingerprint generation
The PMapper is a command line tool that can generate pharmacophore fingerprints for molecules.
References
Pharmacophore - Perception, Development, and use in Drug Design; Osman F. Güner (ed), International University Line, La Jolla, California, 2000.