Stereo Analysis - calculating stereo descriptors
This manual gives you a walk-through on how to use the Stereo Analysis module:
Introduction
Stereochemistry involves the study of the relative spatial arrangement of atoms and their transformations. Stereochemistry defines different isomerisms that describe the molecule's behaviour in the space. These isomer groups cover the entire space of organic molecules. We do not give further details on stereochemistry here, instead we refer to Marvin's stereochemistry summary.
It can be important to determine a molecule's stereochemical behaviour. This can be achieved by calculating stereochemical descriptors for the molecule.
ChemAxon's stereo analysis module is able to calculate stereochemical descriptors for a molecule, giving an analysis in terms of the stereochemical properties.
Usage
The stereo analysis functionality is built in different ChemAxon products.
Stereo analysis with cxcalc
The solubility predictor is integrated into the cxcalc command line tool. The command syntax is
cxcalc [general options] [input files/strings] stereoanalysis [stereoanalysis options] [input files/strings]where the stereoanalysis options are the following:
stereoanalysis options: -h --help this help message -T --type stereo descriptor type [tetrahedral | cistrans | axial | atrop] (default: not set)Some examples of how to use the stereo analysis module via cxcalc :
-
Calculating tetrahedral stereo descriptors for the following molecule:
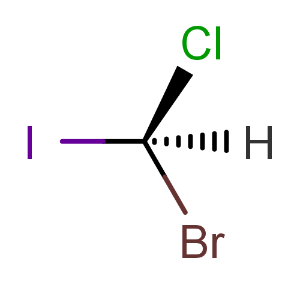
cxcalc stereoanalysis "[H][C@](Cl)(Br)I"The output shows the recognized tetrahedral stereo descriptors:
TETRAHEDRAL [1] - [4, 3, 2, 0] : S -
Calculating cis-trans stereo descriptors:
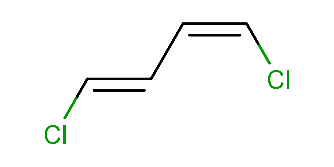
cxcalc stereoanalysis "Cl/C=C\C=C\Cl"The output shows the recognized cis-trans stereo descriptors:
CISTRANS [1, 2] - [0, 3] : ZCISTRANS [3, 4] - [2, 5] : E -
Calculating axial stereo descriptors for a molecule:
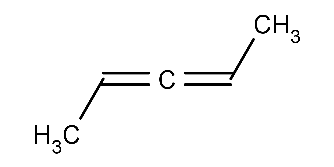
cxcalc stereoanalysis CC=C=CCThe output shows the recognized axial stereo descriptors:
AXIAL [1, 3] - [0, 4] : WIGGLY -
Calculating atrop stereo descriptor for a molecule:
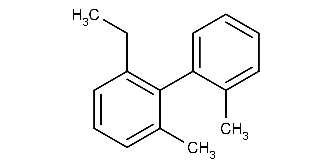
cxcalc stereoanalysis "CCC1=C(C(C)=CC=C1)C1=CC=CC=C1C"The output shows the recognized atrop stereo descriptors:
ATROP [3, 9] - [4, 2, 14, 10] : UNKNOWN -
Calculating all stereo descriptors for a molecule with different stereo features:
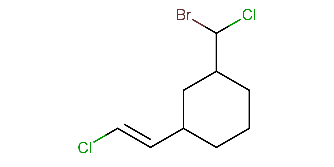
cxcalc stereoanalysis "ClC(Br)C1CCCC(C1)/C=C/Cl"The output shows the recognized stereo descriptors:
TETRAHEDRAL [7] - [9, 8, 6] : UNKNOWNTETRAHEDRAL [3] - [1, 8, 4] : UNKNOWNTETRAHEDRAL [1] - [2, 0, 3] : UNKNOWNCISTRANS [9, 10] - [7, 11] : E -
Listing just the cis-trans stereo descriptors for the previous molecule:
cxcalc stereoanalysis -T cistrans "ClC(Br)C1CCCC(C1)/C=C/Cl"The output is:
CISTRANS [9, 10] - [7, 11] : E
Stereo analysis with Chemical Terms
The stereo analysis module is also integrated into ChemAxon's Chemical Terms language. The stereo descriptors can be calculated by two different functions in Chemical Terms:
-
stereoanalysis(): calculates and returns stereo descriptors of all types for the input molecule.
-
stereoanalysis(T): calculates and returns stereo centers of type T. T can be any stereo function defined in the API.
The following examples show how the functions above can be used with the Chemical Terms evaluator command line tool:
-
Calculating all stereo descriptors for geldanamycin:
evaluate -e "stereoanalysis()" geldanamycinThe output is:
TETRAHEDRAL [26] - [27, 23, 31] : SCISTRANS [18, 20] - [16, 19, 21] : ETETRAHEDRAL [36] - [37, 2, 34] : RTETRAHEDRAL [4] - [3, 6, 5] : RCISTRANS [21, 22] - [20, 23] : ZTETRAHEDRAL [34] - [36, 33, 35] : STETRAHEDRAL [23] - [24, 26, 22] : STETRAHEDRAL [2] - [1, 36, 3] : SCISTRANS [31, 33] - [26, 32, 34] : E -
Calculating only tetrahedral stereo centers for the same molecule:
evaluate -e "stereoanalysis('tetrahedralStereoCenters')" geldanamycinThe output is:
2;4;23;26;34;36 -
Calculating the number of unknown, resolved, tetrahedral stereocenters for the following molecule with the count() function:
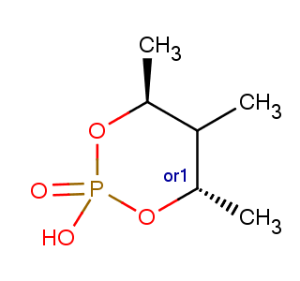
evaluate -e "count(stereoanalysis('unknownResolvedStereoCenters'))" "C[C@@H]1OP(O)(=O)O[C@@H](C)C1C |o1:7,r|" -
Finding only the carbon tetrahedral stereocenters can be done using the filter() function for the same molecule:
evaluate -e "filter(stereoanalysis('tetrahedralStereoCenters'), \"match('C')\")" "C[C@@H]1OP(O)(=O)O[C@@H](C)C1C |o1:7,r|"