Structure Searching
Contents
Database Searching
To assist searching structures in a database, JChem provides the chemaxon.jchem.db.JChemSearch JavaBean. The following search types are supported:
Using a connection object ( ConnectionHandler ) passed by the caller method, JChemSearch retrieves all structures that match the search criteria from the given structure table and returns their cd_id values in an int array.
Oracle users may also use JChem Cartridge for Oracle to perform search and other operations via SQL commands.
Comments:
-
Query molecules may contain query atoms and bonds. For full details, see the JChem Query Guide.
-
Aromatic systems are recognized by JChem even if they are entered with single and double bonds. However the ring will not be considered aromatic if it contains at least one of the following
-
Any atom
-
Hetero atom
-
Any bond
-
Atom list containing at least one atom that excludes aromaticity
-
Negative atom list
-
Aliphatic query property (A)
-
-
JChem applies Hückel's rule for determining aromaticity. (As a result, unlike some other chemical software, JChem considers Kekule form of pyrrole and other five-membered aromatic rings as aromatic.)
-
Implicit and explicit Hydrogen atoms are handled as expected. When Hydrogen atoms are attached to the query structure, the search recognizes implicit Hydrogen atoms in the molecules of the structure table. (For details, see the relevant section in the Query Guide.)
-
Stereo-isomers are distinguished. See Stereo Notes for more details.
-
Coordinates of atoms are neglected during search, except certain stereo searches (chirality or double bond stereo search).
Defining Queries in Web Applications
It is recommended to apply MarvinSketch as a tool for drawing query structures.
Steps of creating a web page for entering query structures:
-
Include a form with a hidden variable with the page that contains MarvinSketch.
-
On submitting the form call MSketch.getMol(...). For example, if the name of the hidden input variable is "molfile" and you need the structure in Marvin format use this call in JavaScript:
form.molfile.value=document.MSketch.getMol('mrv');(You can also get the structure in other formats, like MDL's Molfile or SMILES, but the Marvin format is recommended as it can represent all molecule and query features that are available in Marvin Sketch. You can find more information about file formats here.)
-
Query the requested form's variable in your servlet or server-side script and submit it to the JChemSearch class
Initializing Search
After creating a JChemSearch object , setting the following properties is necessary:
|
the query structure in Smiles, Molfile, or other format |
|
|
ConnectionHandler |
specifies the connection |
|
the table in the database where the structures are stored |
In addition, various other structure search options can be specified that modify structure search behaviour. For further customization, see the API of the JChemSearch class . Many of these options are detailed in the Substructure Search section.
Example:
JChemSearchOptions searchOptions = new JChemSearchOptions(SearchConstants.SUBSTRUCTURE);
JChemSearch searcher = new JChemSearch();
searcher.setStructureTable(structTableName);
searcher.setQueryStructure("Brc1ccccc1");
searcher.setSearchOptions(searchOptions);
searcher.setConnectionHandler(connectionHandler);</b>
searcher.run();Please, see the SearchUtil.java example in the examples/java/search directory.
Please, see SearchTypesExample.java demonstrating search types.
Duplicate Structure Search
This search type can be used to retrieve the same molecule as the query. It is used to check whether a chemical structure already exists in the database, and also during duplicate filter import. All structural features (atom types, isotopes, stereochemistry, query features, etc.) must be the same for matching, but for example coordinates and dimensionality are usually ignored.
For this search mode there is no search per minute license limitation in JChemBase, these searches are not counted.
Java example: Throwing an exception if a given structure exists.
... // Initialize connection
String mol = "Clc1cccc(Br)c1"; // Query in SMILES/SMARTS, MDL Molfile or other format
String structureTableName = "cduser.structures";
JChemSearch searcher = new JChemSearch(); // Create searcher object
searcher.setQueryStructure(mol);
searcher.setConnectionHandler(connHandler);
searcher.setStructureTable(structureTableName);
JChemSearcOptions searchOptions = new JChemSearchOptions( SearchConstants.DUPLICATE );
searcher.setSearchOptions(searchOptions);
searcher.run();
if (searcher.getResultCount() > 0) {
System.out.println("Structure already exists (cd_id=" + searcher.getResult(0) + ")");
}More example:
-
JSP: <JChem's home>/examples/db_search/update.jsp
Substructure Search
Substructure search finds all structures that contain the query structure as a subgraph. Sometimes not only the chemical subgraph is provided, but certain query features also that further restrict the structure. If special molecular features are present on the query (eg. stereochemistry, charge, etc.), only those targets match which also contain the feature. However, if a feature is missing from the query, it is not required to be missing (by default). For more information, see the JChem Query Guide.
Searching starts with a fast screening phase where query and database fingerprints are compared. If the result of the screening is positive (meaning that a fit is possible) for a database structure, then an atom-by-atom search (ABAS) is also performed. Query structures may contain query atoms and bonds described earlier.
Starting a Search
The initialization of substructure searching is similar to duplicate searching , but the JChemSearchOptions object needs to be created with SearchConstants.SUBSTRUCTURE constant value.
Java example:
... // Initialize connection
String mol = "[*]c1cccc([Cl,Br])c1"; // Query structure
String structureTableName = "cduser.structures";
JChemSearch searcher = new JChemSearch(); // Create searcher object
searcher.setQueryStructure(mol);
searcher.setConnectionHandler(connHandler);
searcher.setStructureTable(structureTableName);
JChemSearcOptions searchOptions = new JChemSearchOptions( SearchConstants.SUBSTRUCTURE );
searcher.setSearchOptions(searchOptions);
searcher.run();More examples:
-
JSP: <JChem's home>/examples/db_search/searching.jsp
-
ASP: <JChem's home>/examples/asp/searching.asp
Running Search in a Separate Thread
Since substructure searching can be time consuming, it is reasonable to create a new thread for the search. If the runmode property of JChemSearch is set to JChemSearch.RUN_MODE_ASYNCH_COMPLETE , then searching runs in a separate thread.
The progress of the search can be checked by the following properties of JChemSearch :
|
checks if searching is still running |
|
|
textual information about the phase of the search process |
|
|
the number of hits found so far |
Java application example:
searcher.setRunMode(JChemSearch.RUN_MODE_ASYNCH_COMPLETE);
searcher.run();
while( searcher.isRunning() ) {
String msg = searcher.getProgressMessage() ;
int count = searcher.getResultCount() ;
... // Displaying
Thread.sleep(1000);
}Please, see the AsyncSearchExample.java example in the examples/java/search directory.
JSP example:
if(searcher.isRunning()) {
%>
<p>Please wait. Searching.....
<p><%= searcher.getProgressMessage() >%
<p>Hits: <%= searcher.getResultCount() %>
<p>
<script LANGUAGE="JavaScript">
<!--
window.setTimeout('window.location.reload(false)', 5000);
//-->
</script>
>%
}More examples:
-
JSP: <JChem's home>/examples/db_search/searching.jsp
-
ASP: <JChem's home>/examples/asp/searching.asp
Retrieving Results
If the resultTableMode property of JChemSearch is set to JChemSearch.NO_RESULT_TABLE , then the following properties can be used for retrieving the results:
|
the number of hits found |
|
|
returns true if the search stopped because the time that passed since the start of the searched had reached the maximum value |
|
|
returns true if the search stopped because the number of hits had reached the maximum value |
|
|
returns the cd_id value of a found compound specified by an index value. |
|
|
if an error occurred during the search these properties provide information about the problem |
The two ways of retrieving the results of the search are:
Retrieving Results from the JChemSearch object
The process of retrieving the results of the search from the JChemSearch object is based on the getHitsAsMolecules(. . .) and the getHitsRgDecomp(. . .) methods. In both cases the same JChemSearch object is needed that was used to run the search.
-
Using the getHitsAsMolecules(...) method
-
Apply the array of cd_id values obtained by a JChemSearch.getResults() or JChemSearch.getResult(int) call.
-
Create and configure a HitColoringAndAlignmentOptions object.
Generally, the following properties are set:coloring
Specifies if substructure hit coloring should be used.
enumerateMarkush
Specifies if markush structures should be hit enumerated according to the query structure.
removeUnusedDef
Specifies whether unused R group definitions should be removed from the results.
alignmentMode
Specifies what form of alignment to use for hit display.
The following values are accepted: ALIGNMENT_OFF (default), ALIGNMENT_ROTATE, ALIGNMENT_PARTIAL_CLEAN
-
If this options object is null, the molecules will be returned in their original form.
-
Create an ArrayList of the data field names that should be returned. If this ArrayList is empty, no values will be returned.
-
Create an empty ArrayList to hold the fetched data field values. The ArrayList will have Object array elements, one for each molecule. Inside each Object array will be the fetched data field values.
-
To display the molecules, use the Molecule array return value.
Java example:JChemSearch searcher = new JChemSearch(); // create searcher object // ... // run search int[] resultIds = searcher.getResults(); HitColoringAndAlignmentOptions displayOptions = new HitColoringAndAlignmentOptions(); displayOptions.setColoringEnabled(hitsShouldBeColored); if (hitsShouldBeRotated) { displayOptions.setAlignmentMode(HitColoringAndAlignmentOptions.ALIGNMENT_ROTATE); } else { displayOptions.setAlignmentMode(HitColoringAndAlignmentOptions.ALIGNMENT_OFF); } // ... List<String> dataFieldNames = new ArrayList<String>(); dataFieldNames.add("cd_id"); dataFieldNames.add("cd_formula"); dataFieldNames.add("cd_molweight"); List<Object[]> dataFieldValues = new ArrayList(); Molecule[] mols = searcher.getHitsAsMolecules(resultIds, displayOptions, dataFieldNames, dataFieldValues); // ...More examples:
-
JSP: <JChem's home>/examples/db_search/searchresults.jsp
-
-
Using the getHitsAsRgDecomp(...) method
-
Apply the array of cd_id values obtained by a JChemSearch.getResults() or JChemSearch.getResult(int) call.
-
Set the attachmentType parameter to one of the ATTACHMENT_... constants of the RGroupDecomposition class (the parameter value in most cases is RGroupDecomposition.ATTACHMENT_POINT).
-
Retrieve results by using methods of RgDecompResults
Java example:JChemSearch searcher = new JChemSearch(); // create searcher object // ... // run search int[] resultIds = searcher.getResults(); RgDecompResults rgdResults = searcher.getHitsAsRgDecomp(resultIds, RGroupDecomposition.ATTACHMENT_POINT); // Decomposition objects corresponding to the targets Decomposition[] decompositions = rgdResults.getDecompositions(); // Covering Markush structure of all hits RgMolecule markush = rgdResults.getHitsAsMarkush(); // R-group decomposition table of hits Molecule[][] rgdTable = rgdResults.getHitsAsTable();
-
Retrieving Results using a SQL statement
The process of retrieving the results of the search from the ResultSet Object:
-
Use SQL statements like
SELECT cd_structure, ... FROM cduser.structures WHERE cd_id=12532 -
Apply the cd_id value obtained by a JChemSearch.getResult(...) call in the condition of the SQL statement
To display the molecule, use cd_structure obtained from the ResultSet.
In the case of web applications
-
Use MarvinView to display the data. You can use
-
MarvinView tables (one applet per page)
-
HTML tables (several MarvinView applets per page)
Use the JavaScript routines in marvin.js to make sure that the pages can be viewed in different browsers, GUI-s (AWT/Swing), and JVM-s (built-in/Java plugin).
-
-
Modify values from the cd_structure column using HTMLTools.convertForJavaScript(...) for inserting into a web page in an applet parameter.
Java example:int[] cdIds = searcher.getResults(); String retrieverSql = "SELECT cd_molweight from " + structTableName + " where cd_id = ?";</b> PreparedStatement ps = connectionHandler.getConnection() .prepareStatement(retrieverSql); try { for (int i = 0; i < cdIds.length; i++) { int cdId = cdIds[i]; ps.setInt(1, cdId); ResultSet rs = ps.executeQuery(); if (rs.next()) { System.out.println("Mass: " + rs.getDouble(1)); } else { ; // has been deleted in the meantime? } } } finally { ps.close(); }Please, see the RetrievingDatabaseFieldsExample.java example in the examples/java/search directory.
Please, see MultipleQueriesExample.java demonstrating two approaches for calculating the intersection of the result of multiple queries.
JSP example for displaying search results on a web page by a JSP script:... Hits: <%= searcher.getResultCount() %> <font size="1"> <%= searcher.isMaxResultCountReached()? " (maximum hits reached)" : "" %> <%= searcher.isMaxTimeReached()? " (maximum time reached)" : "" %> </font> <center> <script LANGUAGE="JavaScript1.1" SRC="../../../marvin/marvin.js"> </script> <script LANGUAGE="JavaScript1.1"> <!-- mview_name="mview"; mview_begin("../../../marvin", "<%= cols*cellWidth+cols-1 %>", "<%= rows*cellHeight+rows-1 %>"); mview_param("rows", "<%= rows %>"); mview_param("cols", "<%= cols %>"); mview_param("navmode", "rot3d"); mview_param("molbg", "#000000"); mview_param("bgcolor", "#e0e0e0"); mview_param("border", "1"); mview_param("animate", "0"); mview_param("layout0", ":4:1:"+ "L:0:0:1:1:w:n:0:10:"+ <%/* ID */%> "L:1:0:1:1:w:n:0:10:"+ <%/* Stock */%> "M:2:0:1:1:c:n:1:10"); <%/* Molecule */%> mview_param("param0", ":"+ "L:11b:"+ "L:10:"+ "M:<%= structureWidth %>:<%= structureHeight %>"); <% /* * Writing data into the cells in the applet */ int cellIndex=0; for(int i=start; i<start+cells; i++) { int id = searcher.getResult(i); /* * SQL statement for retrieving structures */ String sql = "SELECT " + structureTableName + ".cd_id, "+ structureTableName + ".cd_structure, " + stockTableName + ".quantity\n" + "FROM "+ structureTableName + ", " + stockTableName + "\n" + "WHERE " + structureTableName + ".cd_id = " + stockTableName + ".cd_id AND " + structureTableName + ".cd_id = " + id; Statement stmt = con.createStatement(); ResultSet rs = stmt.executeQuery(sql); try { if(rs.next()) { String dbMolfile = new String(DatabaseTools.readBytes(rs, 2),"ASCII"); float dbQuantity = rs.getFloat(3); %> mview_param("cell<%= cellIndex %>", "|ID: <%= id %>"+ "|<%= Math.round(dbQuantity) %> mg"+ "|<%= HTMLTools.convertForJavaScript( dbMolfile) %>"); <% } } finally { rs.close(); stmt.close(); } cellIndex++; } %> mview_end(); //--> </script> </center> ...Click here to see the live result of the above code.
More JSP examples:
-
<JChem's home>/examples/db_search/searchresults.jsp
-
<JChem's home>/examples/simple_db_search/searchresults.jsp
To store the results in a table, the name of the table should be specified by the resultTable property of JChemSearch , and also the resultTableMode property should be set to either JChemSearch.CREATE_OR_REPLACE_RESULT_TABLE or JChemSearch.APPEND_TO_RESULT_TABLE .
Retrieving hits as soon as they are found
If the runmode property of JChemSearch is set to JChemSearch.RUN_MODE_ASYNCH_PROGRESSIVE , then searching runs in a separate thread and hits can be retrieved as soon as they are found. Note that this mode does not support any ordering:
List<int[]> hitsByPages = new ArrayList<int[]>();
int[] nextPage = new int[NUMBER_OF_HITS_PER_PAGE];
int idxForNextPage = 0;
*searcher.setOrder(JChemSearch.NO_ORDERING)* ;
*searcher.setRunMode(JChemSearch.RUN_MODE_ASYNCH_PROGRESSIVE)* ;
*searcher.run()* ;
while ( *searcher.hasMoreHits()* ) {
nextPage[idxForNextPage++] = *searcher.getNextHit()* ;
if (idxForNextPage == NUMBER_OF_HITS_PER_PAGE) {
synchronized (hitsByPages) {
hitsByPages.add(nextPage);
hitsByPages.notifyAll(); // notify any who may be in wait for the next page
nextPage = new int[NUMBER_OF_HITS_PER_PAGE];
idxForNextPage = 0;
}
}
}
// Hits for the last page if any
if (idxForNextPage > 0) {
int[] lastPage = new int[idxForNextPage];
System.arrayCopy(nextPage, 0, lastPage, 0, idxForNextPage);
synchronized (hitsByPages) {
hitsByPages.add(lastPage);
hitsByPages.notifyAll(); // notify any who may be in wait for the next page
}
}Caching Structures
To boost the speed of substructure searching, JChem caches fingerprints and structures in the searcher application's memory. For more information, see the JChem database concepts section.
Combining SQL queries with Structure Searching
Many times structure information is only one of several conditions that a complex query has to check. In those cases structure searches should be combined with SQL queries.
Example: Suppose that quantities on stock are stored in a table different from the structure table. We are querying compounds that contain a given substructure and their quantity on stock is not less than a given value.
Two ways of performing the combined query:
-
Structure Searching Followed by SQL Query
-
Initialize a JChemSearch object.
-
Instruct JChemSearch to save the cd_id values of found compounds: set the name of the result table using the setResultsTable method.
-
Run structure searching.
-
Query the join of the structure table, the result table, and the stock table using an SQL SELECT statement. (See the syntax of SELECT in the documentation of your database engine.) Example:
SELECT cd_structure, quantity FROM hits, structures, stock WHERE hits.cd_id=structures.cd_id AND stock.cd_id=structures.cd_id AND stock.quantity < 2
-
-
SQL Query Followed by Structure Searching
An arbitrary SQL query can be specified as a filter for the filterQuery property. The (first) result column should contain the allowed cd_id values.
Java example:JChemSearchOptions jcSearchOptions = new JChemSearchOptions(SearchConstants.SUBSTRUCTURE); jcSearchOptions.setFilterQuery("SELECT cd_id FROM " + stockTableName + " WHERE quantity < 2");Please, see the SearchWithFilterQueryExample.java example in the examples/java/search directory.
Calculated columns
Databases of chemical structures can contain various calculated values. These are specified upon table creation using Chemical Terms expressions and their value is calculated during database import. Please, see how calculated columns are stored in JChem tables. While executing database search these fields can be considered. Assuming that the table "search_example" exists with the columns logp, rtbl_bnd_cnt and pka_ac_2 a search can be executed as follows:
String[] columns = { "logp", "rtbl_bnd_cnt", "pka_ac_2" };
double[] thresholds = { 3.85, 3, 18 };
for (int i = 0; i < columns.length; i++) {
searchOptions.setFilterQuery("SELECT cd_id FROM " + tableName
+ " WHERE " + columns[i] + ">" + thresholds[i]);
jcs.setSearchOptions(searchOptions);
jcs.run();
... // handling results
}
Please, see how to create calculated columns with JChem Manager.
Using command line tool the following command performs the same operation:
jcman c search_example
--coldefs ", logp float8, rtbl_bnd_cnt float8, pka_ac_2 float8"
--ctcolcfg "logp=logp(); rtbl_bnd_cnt=rotatableBondCount()>4; pka_ac_2=pKa(\"acidic\",\"2\")"To an existing table, where the appropriate columns have been created, calculated columns can be added using jcman tool:
jcman m search_example
--add-ctcolcfg "logp=logp(); rtbl_bnd_cnt=rotatableBondCount()>4; pka_ac_2=pKa(\"acidic\",\"2\")"Please, see CalculatedColumnsSearchExample.java demonstrating the usage of calculated columns during search.
Chemical Terms filtering
Calculated values do not need to be stored in a database field. If they should be used for a temporary filtering, they may be calculated "on the fly". These are specified for database search using the setChemTermsFilter option:
searchOptions.setChemTermsFilter("pka(h(0))> 2");Please, see SortedSearchExample.java demonstrating hits ordering.
Please, see SearchWithFilterQueryExample.java demonstrating how to filter search results based on other (possibly) database tables.
Setting More Properties
There are several other properties that modify the behavior of JChemSearch , like
|
The maximum number of molecules that can be found by the search. |
|
|
The maximum amount of time in milliseconds, which is available for searching. |
|
|
stringToAppend |
A string (like an ORDER BY sub-expression) to be appended to the SQL expression used for screening and retrieving rows from the structure table. |
|
infoToStdError |
If set to true, information useful for testing will be written in the servlet server's error log file. |
|
Specifies the order of the result. Java example: Please, see the SortedSearchExample.java example in the examples/java/search directory. |
Superstructure Search
Superstructure search finds all molecules where the query is superstructure of the target. It can be invoked in a similar fashion as Substructure search. In case of superstructure search note that except query tables the default standardization removes explicit hydrogens (see query guide) This has the effect that structures in the database are used without their explicit hydrogens as queries in these cases.
Set search type to SearchConstants.SUPERSTRUCTURE .
Full Structure Search
A full structure search finds molecules that are equal (in size) to the query structure. (No additional fragments or heavy atoms are allowed.) Molecular features (by default) are evaluated the same way as described above for substructure search.
For this search type, the JChemSearchOptions object needs to be created with the SearchConstants.FULL value.
Full fragment Search
Full fragment search is between substructure and full search: the query must fully match to a whole fragment of the target. Other fragments may be present in the target, they are ignored. This search type is useful to perform a "Full structure search" that ignores salts or solvents beside the main structure in the target.
For this search type, the JChemSearchOptions object needs to be created with the SearchConstants.FULL_FRAGMENT value.
Similarity Search
Similarity searching finds molecules that are similar to the query structure. Per default the search uses Tanimoto coefficient . Tanimoto coefficient has two arguments:
-
the fingerprint of the query structure
-
the fingerprint of the molecule in the database The dissimilarity formula contains the Tanimoto coefficient and measures how dissimilar the two molecules are from each other. The formula is defined below:
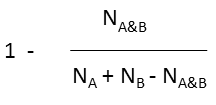
where N A and N B are the number of bits set in thefingerprint of molecule A and B, respectively, N A&B is the number of bits that are set in both fingerprints.
Other dissimilarity metrics can be set by the setDissimilarityMetric function. Possible values:
-
Tanimoto - default
-
Tversky
-
Euclidean
-
Normalized_euclidean
-
Dice
Substructure
Superstucture Tversky index can have two weights, those values are also set in the String parameter of the function using the comma as separator. For example,JChemSearchOptions searchOptions = new JChemSearchOptions(SearchConstants.SIMILARITY); searchOptions.setDissimilarityMetric("Tversky,0.3,0.6");The dissimilarity threshold is a number between 0 and 1, which specifies a cutoff limit in the similarity calculation. If the dissimilarity value is less than the threshold, then the query structure and the given database structure are considered similar.
See more details on fingerprints in the section Parameters for Generating Chemical Hashed Fingerprints.
Similarity searching should be used the same way as substructure searching . To enable similarity searching, the JChemSearchOptions object need to be created with the SearchConstants.SIMILARITY value. If the order property is set to JChemSearch.ORDERING_BY_ID_IR_SIMILARITY (which is the default), then the hits returned by the getResult() method will be sorted in increasing order of dissimilarity.
The dissimilarity threshold is set on JChemSearchOptions with this function:
|
setDissimilarityThreshold(float dissimilarityThreshold) |
Sets the dissimilarity threshold. Expects a float value between 0 and 1. A lower threshold results in hits that are more similar to the query structure. |
The dissimilarity values predicted in the similarity calculation are retrieved with the JChemSearch instance with this function:
|
getDissimilarity(int index) |
Returns the predicted dissimilarity value for the hit corresponding to the given index. |
Java example:
JChemSearch searcher = new JChemSearch(); // Create searcher object
searcher.setQueryStructure(mol);
searcher.setConnectionHandler(connHandler);
searcher.setStructureTable(structureTableName);
JChemSearcOptions searchOptions = new JChemSearchOptions(JChemSearch.SIMILARITY);
searchOptions.setDissimilarityThreshold(0.2f);
searcher.setSearchOptions(searchOptions);
searcher.run();
...
for (int i = 0; i < searcher.getResultCount(); i++) {
float similarity = searcher.getDissimilarity(i);
// ...
}More examples:
-
JSP: <JChem's home>/examples/db_search/searching.jsp
-
ASP: <JChem's home>/examples/asp/searching.asp
If a result table is generated during a similarity search, then the table will contain both the cd_id and the calculated similarity values.
Similarity Searching With Molecular Descriptors
Users can open up new ways of similarity searching by using a number of built-in molecular descriptor types other than the default chemical hashed fingerprints. There are a number of built-in molecular descriptors available, including CF, PF, Burden eigenvalue descriptor (or BCUTTM) and various scalar descriptors.
The following example shows how simple it is to setup molecular descriptors for your compound library. The first command creates a table called compound_library and the second command adds the molecules from an sdf file. The third command uses the 'c' option to create the molecular descriptor with the name of the structure table set by the -a flag, and the chemicalfingerprint descriptor type set by the -k flag. The command omits the database login information that was stored previously with the -s option. See the jcman command options and the GenerateMD command options for more information. Creating and assigning molecular descriptors to database structure tables is discussed with the GenerateMD command.
jcman c compound_library
jcman a compound_library my_compound_group.sdf
generatemd c -a *compound_library* -k CF chemical_fingerprint Below is an example that runs the similarity search with the new chemical fingerprint. The molecular descriptor name, chemical_fingerprint , is set as a search option and the similarity search is run normally:
JChemSearch searcher = new JChemSearch(); // Create searcher object
searcher.setQueryStructure(mol);
searcher.setConnectionHandler(connHandler);
searcher.setStructureTable("compound_library");
JChemSearcOptions searchOptions = new JChemSearchOptions(JChemSearch.SIMILARITY);
searchOptions.setDescriptorName("chemical_fingerprint");
searchOptions.setDissimilarityThreshold(0.2f);
searcher.setSearchOptions(searchOptions);
searcher.run();Molecular Descriptor Configuration Options
Application end-users may need further information about the molecular descriptors to select an appropriate molecular descriptor for their search. Application developers can extract this information from the database and display it to the end-user to help with selection. In this Java example, a MDTableHandler is created using the database connection and the name of the structure table. The MDTableHandler provides access to the Molecular Descriptors and the embedded configurations, metrics, and default dissimilarity thresholds.
// Start with database connection handler and name of the structure table
MDTableHandler mdth = new MDTableHandler(connectionHandler, structureTableName);
String[] descriptorIds = mdth.getMolecularDescriptors();
for (int x = 0; x < descriptorIds.length; x++){
String mdName = descriptorIds[x];
MolecularDescriptor descriptor = mdth.createMD(mdName);
// Ge descriptor names
String descriptorName = descriptor.getName();
// Get descriptor comments
String descriptorComment = mdth.getMDComment(mdName);
// Get available metrics for each configuration
String[] configNames = mdth.getMDConfigs(mdName);
for (int i = 0; i < configNames.length; i++) {
MolecularDescriptor tempDescr = (MolecularDescriptor) descriptor.clone();
String config = mdth.getMDConfig(mdName,configNames[i]);
tempDesc.setScreeningConfiguration(config);
// Get metric name
String metricName = tempDesc.getMetricName();
// Get default thresholds
String defaultThreshold = tempDesc.getThreshold();
// ...
}
// Display code can go here
// ...
}After selecting a molecular descriptor and other desired parameters, such as the descriptor configuration and the metric, the custom molecular descriptor name is set as a search option and the similarity search is run as normal. If the descriptor name, configuration or metric is omitted, a stored default value is used.
JChemSearch searcher = new JChemSearch(); // Create searcher object
searcher.setQueryStructure(mol);
searcher.setConnectionHandler(connHandler);
searcher.setStructureTable(structureTableName);
JChemSearcOptions searchOptions = new JChemSearchOptions(JChemSearch.SIMILARITY);
searchOptions.setDescriptorName(selectedDescriptor);
searchOptions.setDescriptorConfig(selectedConfig);
searchOptions.setDissimilarityMetric(selectedMetric);
searchOptions.setDissimilarityThreshold(0.8f);
searcher.setSearchOptions(searchOptions);
searcher.run();More examples:
Customizing the Molecular Descriptor
In addition, a cheminformatics expert can generate and fine tune a custom made molecular descriptor. Further information on generating the custom molecular descriptors can be found here .
Please, see SimilaritySearchExample.java demonstrating how descriptors are generated and similarity searches executed on them.
Formula search
Formula search is applicable for finding molecules in JChem structure tables using the cd_formula field. Formula search can be combined with other database searching methods, that is with duplicate structure search, substructure search, full structure search, full fragment search, superstructure search, and similarity search.
Types of formula search are the following:
-
Exact Search
-
Exact Subformula Search
-
Subformula Search For more detailed description about formula search functionalities go to query guide.
Example:
JChemSearchOptions searchOptions = new JChemSearchOptions(SearchConstants.SUBSTRUCTURE);
searchOptions.setFormulaSearchQuery("C3-H10-");
searchOptions.setFormulaSearchType(FormulaSearch.EXACT);
JChemSearch searcher = new JChemSearch();
searcher.setConnectionHandler(connHandler);
searcher.setStructureTable(tableName);
searcher.setQueryStructure(""); // note that no structural query is used
searcher.setSearchOptions(searchOptions);
searcher.run();To perform formula search in memory see Sophisticated Formula Search .
Search Access Level
The maximum number of substructure and similarity searches allowed by the system per minute is determined by the license key entered using JChemManager. If no license key has been specified, then the program is in demo mode that allows one search per minute.
If a query is started when the number of searches has exceeded the quota, JChemSearch throws MaxSearchFrequencyExceededException. It is recommended to catch this exception and display a friendly message advising the user to try searching later. If this exception occurs frequently, please contact ChemAxon |mailto:'+ 'sales''@''chemaxon.com] and request a license key allowing more searches. Click here to display a table that helps you to determine the access level that suits your needs. For further details see a href="jchemtest_dev_dbconcepts_index#fingerprints"> Query guide The maximum number of substructure and similarity searches allowed by the system per minute is determined by the license key entered using JChemManager. If no license key has been specified, then the program is in demo mode that allows one search per minute.
Structure Searching in memory and flat files
The searching of in-memory molecules ( chemaxon.struc.Molecule objects) can be performed by the use of chemaxon.sss.search.MolSearch or chemaxon.sss.search.StandardizedMolSearch classes.
Files and strings
If the files to be searchable are only available in a molecular file format in a string or stored in the file system, they have to be imported into Molecule objects by the use of chemaxon.formats.MolImporter or chemaxon.util.MolHandler classes. The code example at the MolSearch API description shows examples for the use of both classes.
Various Java examples for importing molecules using JChem API are available in Java and HTML format.
An easy to use command line tool for searching and comparing molecules in files, databases or given as SMILES strings is jcsearch.
Usage of MolSearch and StandardizedMolSearch
A search object of these classes compares two Molecule objects (a query and a target) to each other. Usually a MolSearch object is used in the following scenario:
MolSearch searcher = new MolSearch(); // create search object
queryMol.aromatize(); // aromatize the query structure
searcher.setQuery(queryMol); // set the query to search
targetMol.aromatize(); // aromatize the target molecule
searcher.setTarget(targetMol); // set the target molecule to search
// Create search options object and set the search type
MolSearchOptions searchOptions =
new MolSearchOptions(SearchConstants.SUBSTRUCTURE);
// Set other search options here.
// For more information, see MolSearchOptions and its superclass, SearchOptions.
// ...
// Set the search options to the searcher
searcher.setSearchOptions(searchOptions);
// Perform search and obtain results
// ... Another way of comparing molecules is using StandardizedMolSearch is a descendant of MolSearch and disposes all its methods. StandardizedMolSearch calls standardization during the search functions automatically. If no standardization configuration is given, the default standardization is applied, which consists of an aromatization step. Hence using StandardizedMolSearch no initial aromatization of the input molecules is required. Therefore we suggest using the StandardizedMolSearch class.
|
Search operation |
Description |
|
ms.isMatching() |
The most efficient way to decide whether there is a match between query and target. |
|
ms.findAll() |
Looks for all occurrences(matching) of query in target. Returns an array containing the matches as arrays (int[][]) or null if there are no hits. The match arrays(int[]) contain the atom indexes of the target atoms that match the query atoms (in the order of the appropriate query atoms). |
|
ms.findFirst() and consecutive ms.findNext() calls |
Same as findAll() above, but return individual match arrays one by one. findFirst() re-initializes the search object, and starts returning matches from the start. Both return null, if there are no more hits to return. |
|
ms.getMatchCount() |
Returns the number of matchings between query and target. |
Please, see MemorySearchExample.java demonstrating the usage of MolSearch class.
Please, see StandardizedMolSearchExample.java demonstrating the usage of StandardizedMolSearch class.
For further information, see the following resources:
-
Structural Search Using ChemAxon Tools (.ppt or .pdf poster version)
-
Power search: How to tune search for efficiency and performance - 2006 UGM presentation
Duplicate search
There are several ways for searching for duplicates in a file. First you have to import the file as described in files and strings. Then you can search the read array of Molecule objects in the following ways.
-
Make a double loop through all the molecules and compare them using MolSearch. (see example code )
-
Generate unique SMILES representation of the Molecule objects and compare these Strings. For generating unique SMILES strings see: smiles export
For the comparison, an efficient data structure can be used (e.g. java.util.HashSet).
Code example:List<Molecule> mols; // ... Set<String> smilesSet = new HashSet<String>(); for (int i = 0; i < mols.size(); i++) { // Create unique SMILES representation String smiles = MolExporter.exportToFormat(mols.get(i), "smiles:u"); // Check if the same unique SMILES has already been found if (!smilesSet.contains(smiles)) { smilesSet.add(smiles); } else { // duplicate structure is found } }See the full source code here.
-
Generate the molecules' hash codes and compare them. These hash codes are equivalent if the molecules are the same, but the equivalence of the code doesn't necessarily imply that the molecules are the same. This should be verified using structure searching. Thus this way of comparison is efficient if the number of duplicates is relatively small compared to the number of molecules.
Code example:StandardizedMolSearch searcher = new StandardizedMolSearch(); HashCode hc = new HashCode(); // Generate hash codes int[] codes = new int[mols.size()]; for (int i = 0; i < mols.size(); i++) { codes[i] = hc.getHashCode(mols.get(i)); } // Search for duplicates based on hash code comparison and subsequent searching for (int q = 0; q < mols.size(); q++) { for (int t = 0; t < q; t++) { if (codes[q] == codes[t]) { // If hash-codes are equal, check with MolSearch searcher.setQuery(mols.get(q)); searcher.setTarget(mols.get(t)); if (searcher.isMatching()) { // duplicate structure is found break; } } } }See the full source code here.
Sophisticated Formula Search
Formula search finds molecules that have matching chemical formulas to a given query. In order to use this class the formulas of the molecules need to be obtained. This can be accessed through the cd_formula column or any other used defined/calculated column of a database which contains a valid chemical formula. The ElementalAnalyser class can be used to obtain the formula or dot disconnected formula of a molecule. A method which would match two molecules by formula:
FormulaSearch searcher = new FormulaSearch() searcher.setSearchType(FormulaSearch.EXACT); searcher.setQuery("C3H7NO"); searcher.setTarget(targetFormula); return searcher.isMatching();
Stereo Notes
Tetrahedral centers and double bond stereo configurations are recognized during searching. The information applied by JChem for stereo recognition is
-
in 0D (Smiles): explicit stereochemical notation using the @, @@, /, \ symbols in the Smiles string
-
in 2D: wedge bonds (in the case of chiral centers), enhanced stereo labels and bond angles (for double bond E/Z information)
in 3D: coordinates JChemSearch handles all reasonable structures appropriately. When the query structure is specified in MDL Molfile or Marvin mrv formats for JChemSearch , and E/Z stereoisomers are searched, the stereo search attribute (or stereo care flag) of the bonds has to be set. See the relevant section of the Query Guide . Furthermore, only the following formats supports the enhanced stereo configuration of stereocenters: MDL extended (V3000) formats, Marvin mrv, ChemAxon extended smiles/smarts. More details on these are available at the following sources: -
JChem User's Guide: JChem Query Guide, Stereochemistry section
-
File format informations: Marvin mrv format, MDL mol formats, ChemAxon extended smiles and smarts formats.
-
the documentation of the MolBond class
-
MDL's CTFILE Formats documentation The database structures may be imported in MDL SDfile, Molfile or Daylight SMILES format. The JChem Class Library provides classes and applications for interconverting between different formats (e.g. see the chemaxon.util.MolHandler object).